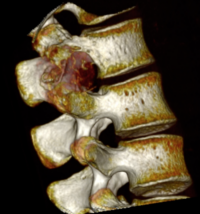
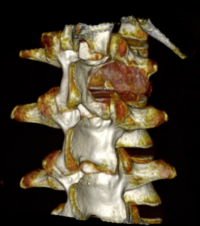
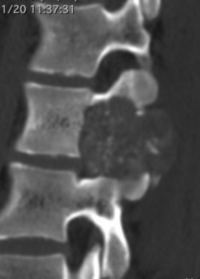
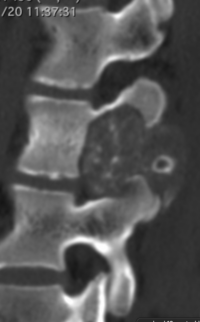
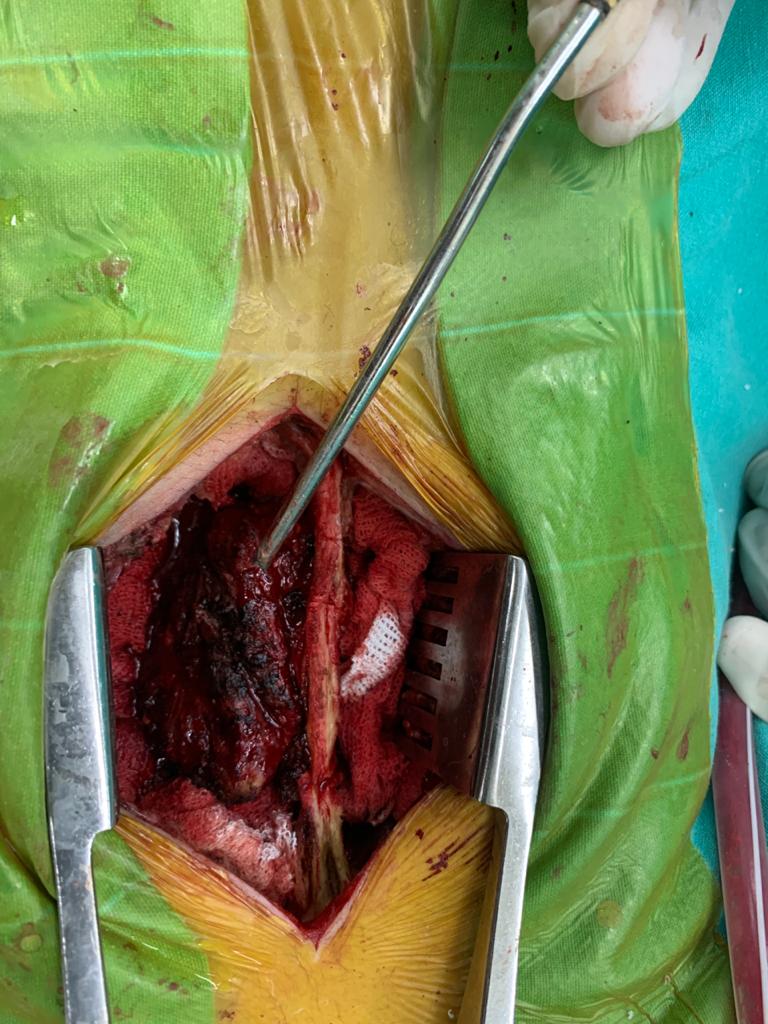
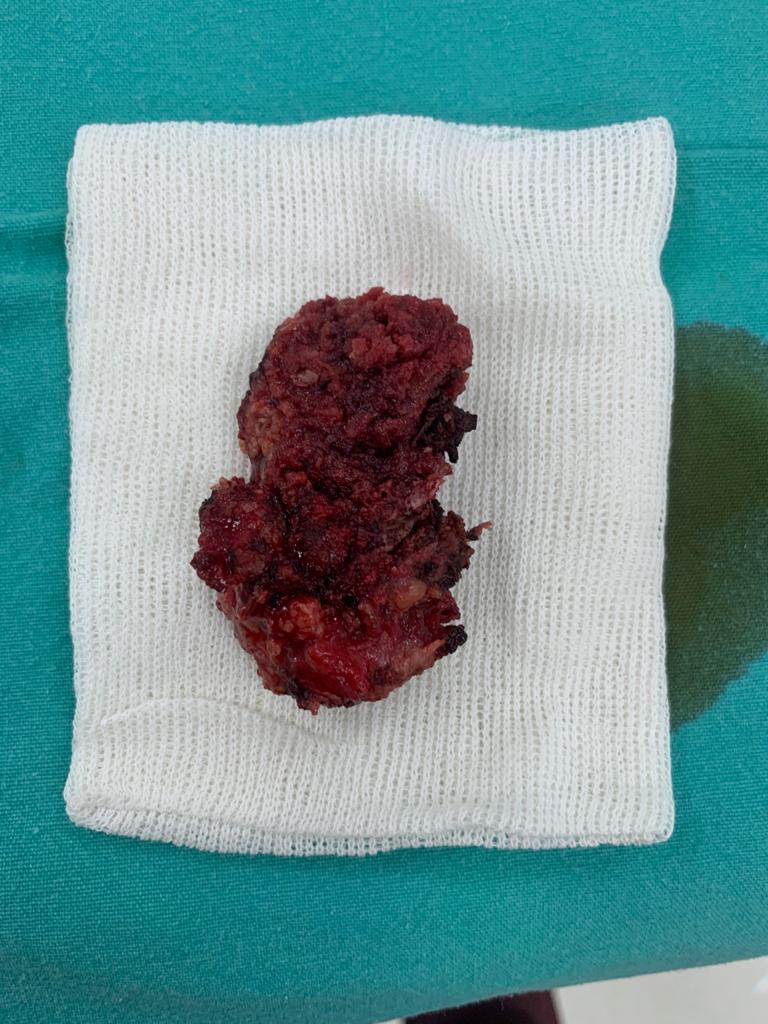
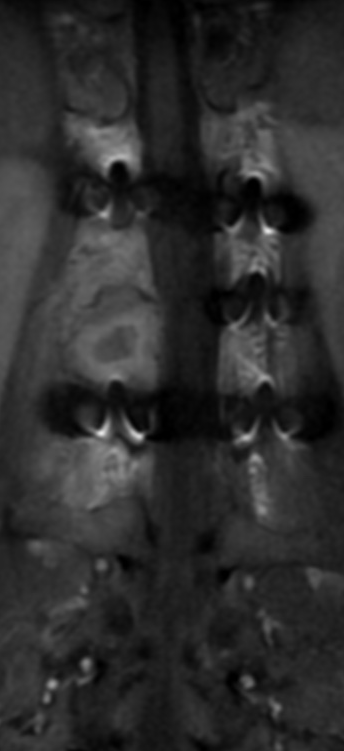
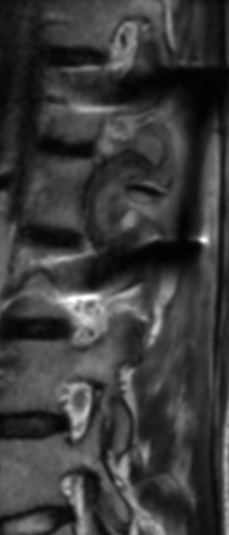
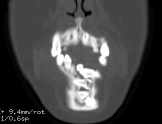
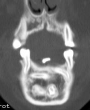
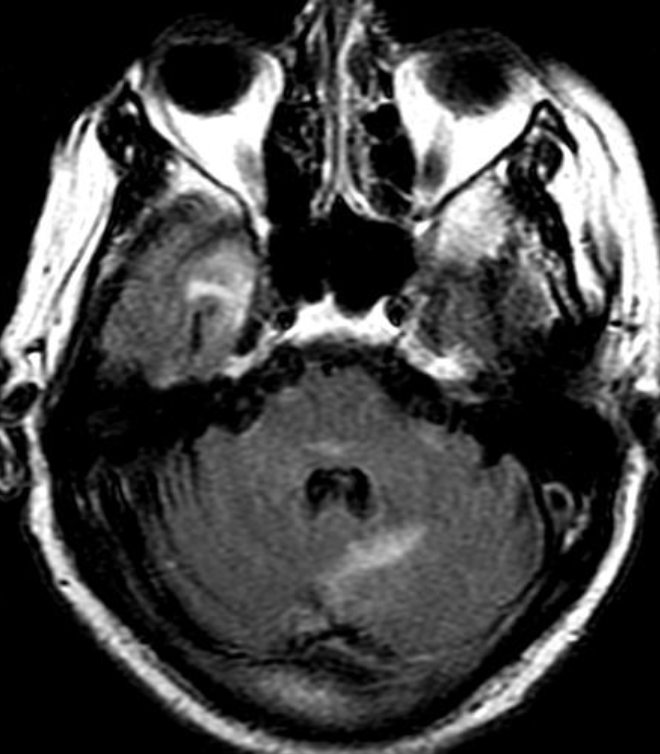
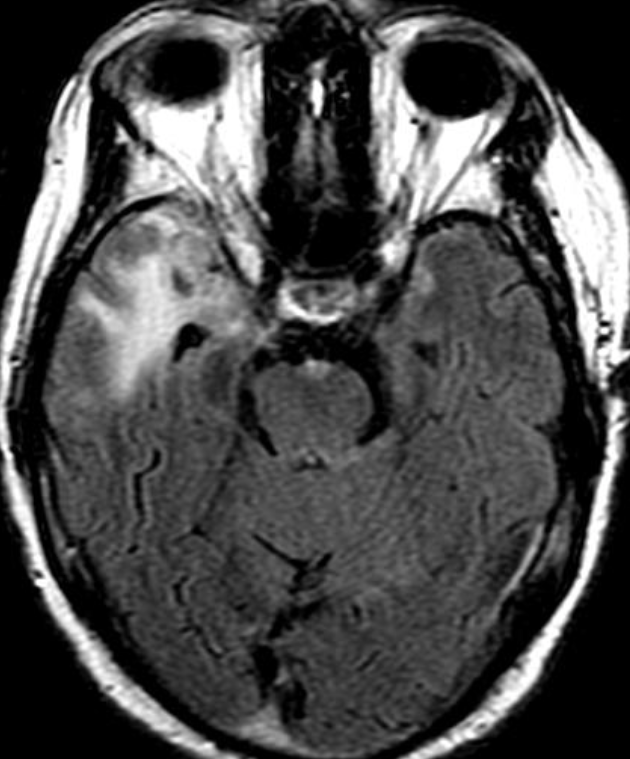
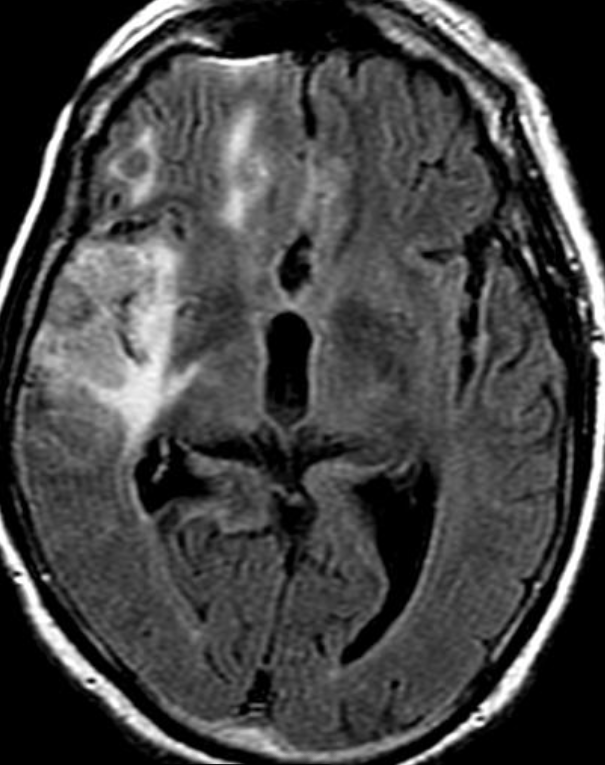
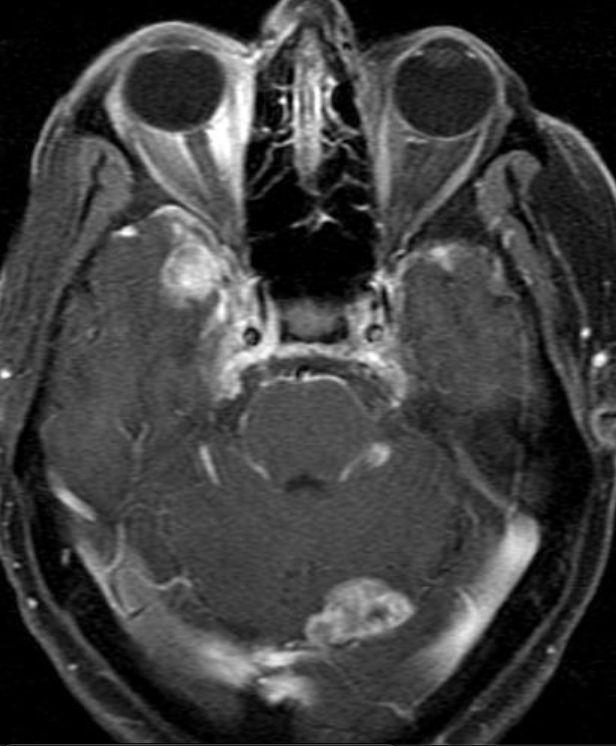
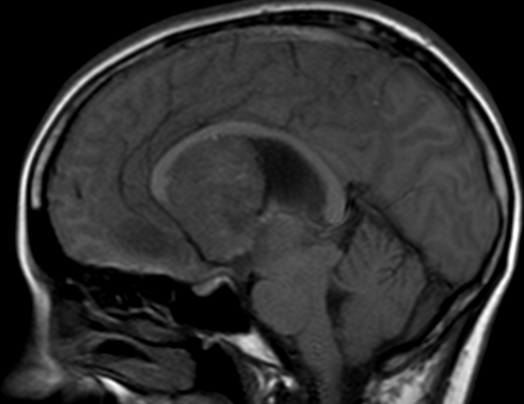
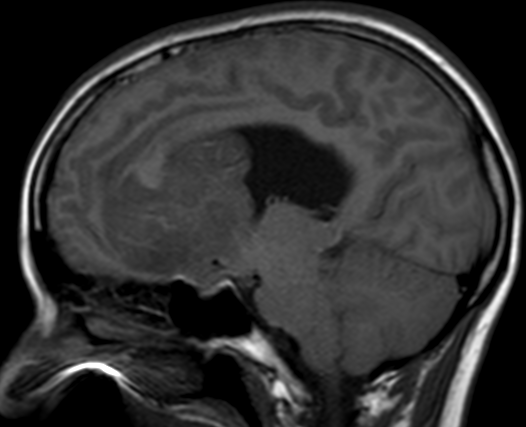
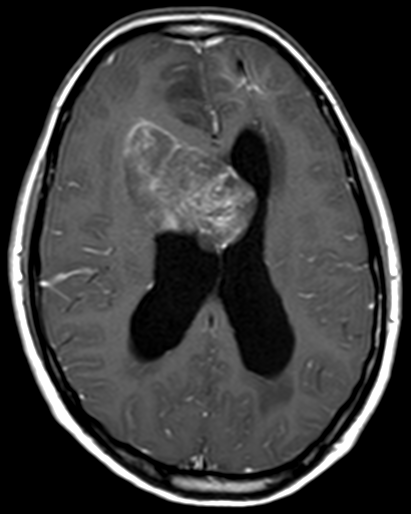
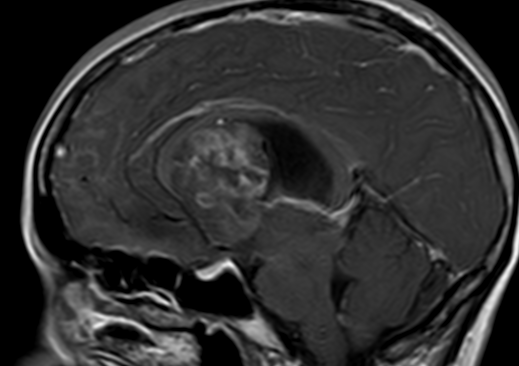
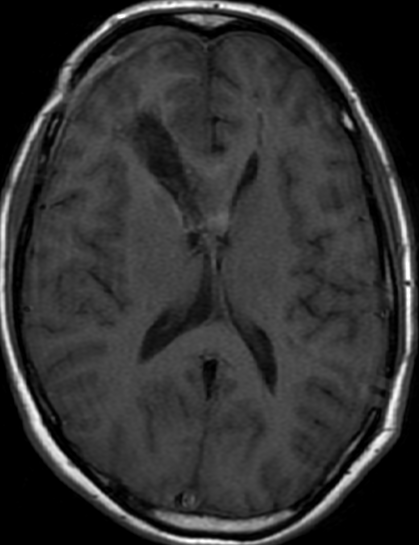
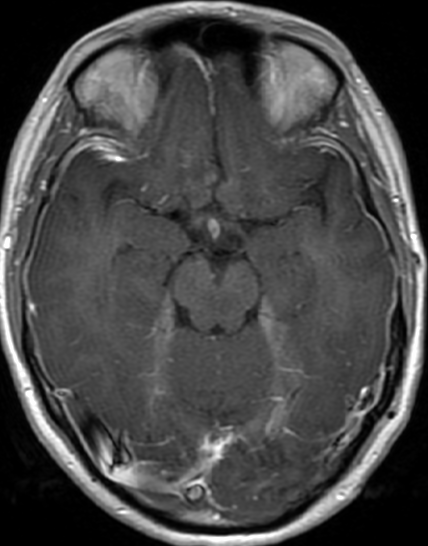
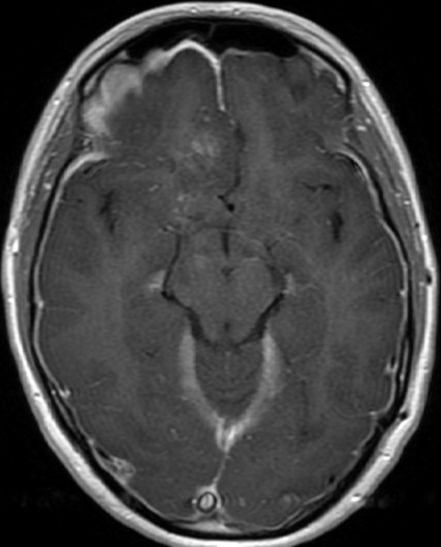
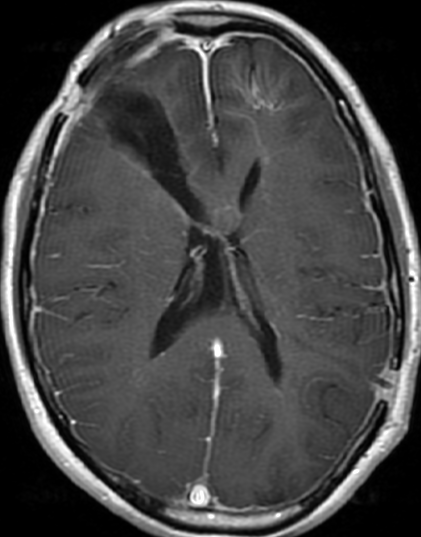
OBs are very rare and constitute about 1% of all primary bony tumors. These tumors tend to involve long bones and vertebral column. OBs are rare primary bony tumors constituting only 1% of all cases.
In 1984, a borderline osteoblastic tumor entity called “aggressive osteoblastoma” was introduced by Dorfman and Weiss and was found to be associated with a high recurrence rate and potential for malignant transformation. Despite being benign tumors, osteoblastomas often cause pronounced bone destruction, soft tissue infiltration, and epidural extension. They usually behave aggressively with extensive uncontrollable local recurrence, and even malignant transformation with metastatic disease has been reported
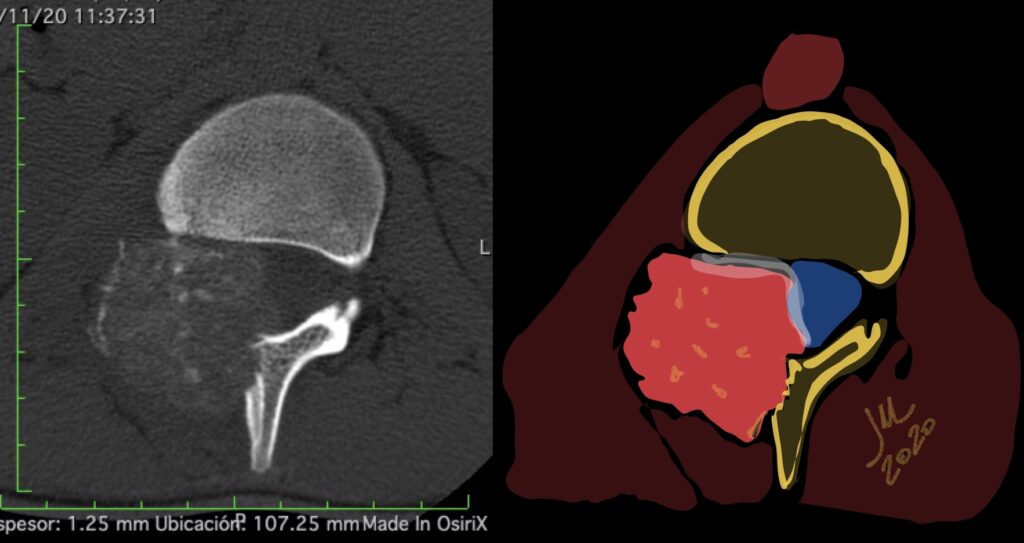
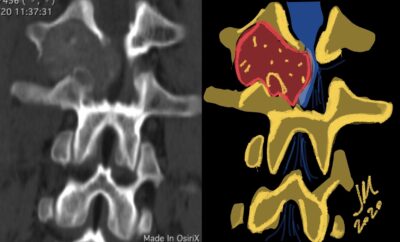
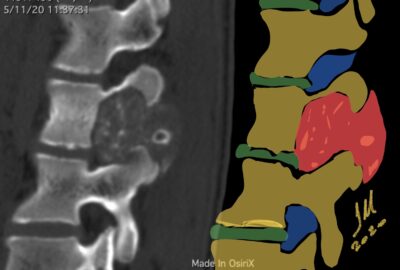
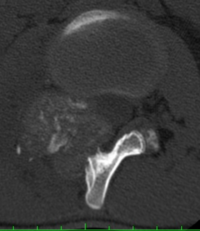
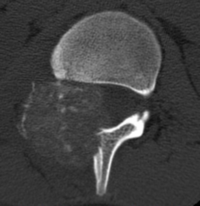
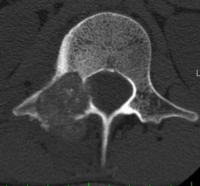
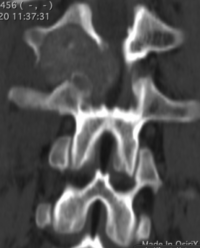
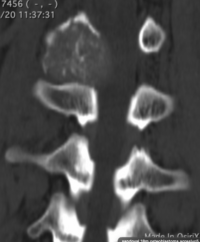
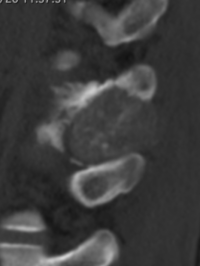
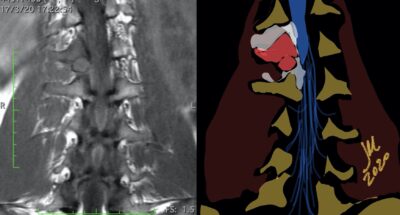
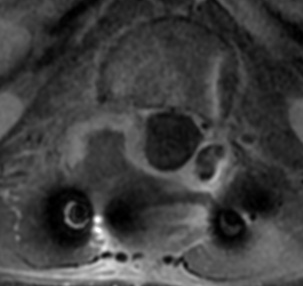
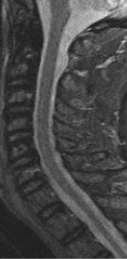
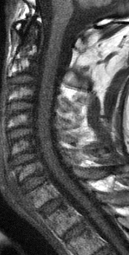
CT appearance of spinal osteoblastoma is a lytic expansile lesion with a shell of sclerosis, and soft tissue masses can be observed around the lesion.
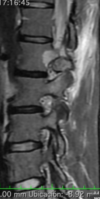
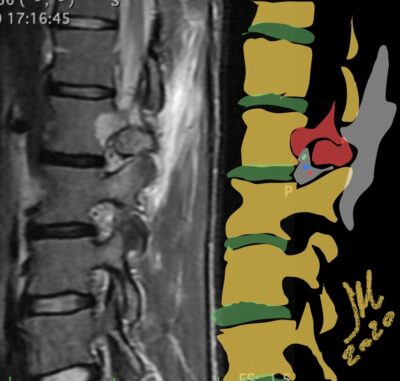
The imaging features of spinal osteoblastoma on MRI are considered nonspecific. These lesions often display a low to isointense signal on T1-weighted MRI, whereas T2-weighted MRI demonstrates an intermediate- to high-intensity signal due to matrix calcification and intense enhancement representing the highly vascular nature of such lesions
Notably, however, this radiographic appearance may lead to overestimation of the limits of the tumor and can be somewhat confusing to orthopedic surgeons and radiologists, who may interpret these tumors as other clinical entities such as Ewing’s sarcoma or lymphoma
flare phenomenon
flare phenomenon” was described by Crim et al. in 1990. Specifically, these neoplasms often cause a diffuse, reactive prostaglandin (PG) – mediated inflammatory infiltrate within adjacent vertebrae surrounding paraspinal soft tissues. In contrast, for aggressive osteoblastoma in the spine, the tumor may be an expansile lesion with a multitude of small calcifications, a prominently clerotic rim, and paravertebral and epidural extensions and may also radiographically mimic aneurysmal bone cysts, osteosarcomas, or bone metastases.
flare phenomenon
Another essential diagnostic tool is bone scintigraphy, which is the most sensitive radiographic scan described in the literature for osteoblastoma and typically shows increased radiotracer uptake, indicating increased osseous turnover
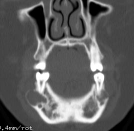
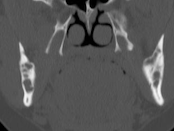
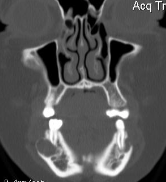
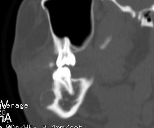
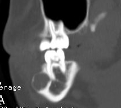
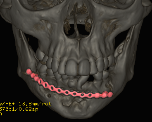
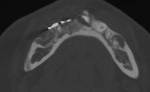
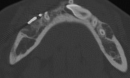
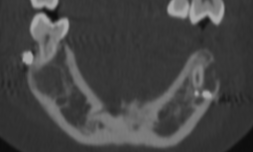
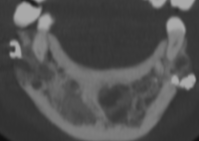
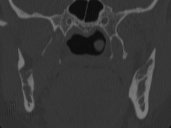
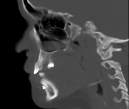
Cherubism is a rare osseous disorder of children and adolescents. Although the radiologic characteristics of cherubism are not pathognomonic, the diagnosis is strongly suggested by bilateral relatively symmetric jaw involvement that is limited to the maxilla and mandible. Imaging typically shows expansile remodeling of the involved bones, thinning of the cortexes, and multilocular radiolucencies with a coarse trabecular pattern.
Cherubism has been described as a subtype of fibrous dysplasia, specifically a hereditary craniofacial fibrous dysplasia, because of the radiographic similarities between the conditions. Recent genetic analysis, however, has shown them to be separate entities. The genetic basis for cherubism was identified in 1999, when the gene responsible for it was mapped to chromosome 4p16.3.
16F. 1 TC
Although the radiologic and histologic characteristics of cherubism are not pathognomonic, the overall morphologic features are characteristic and consistent among cases; these features therefore allow an accurate prospective diagnosis in the appropriate clinical setting.
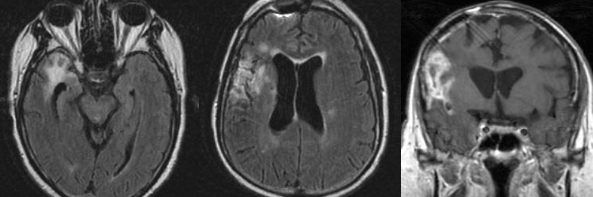
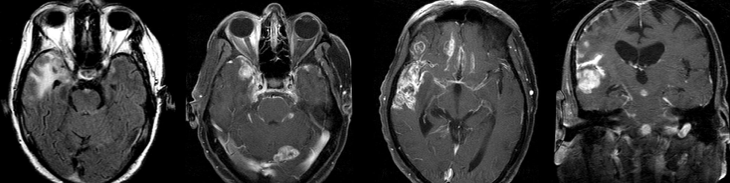
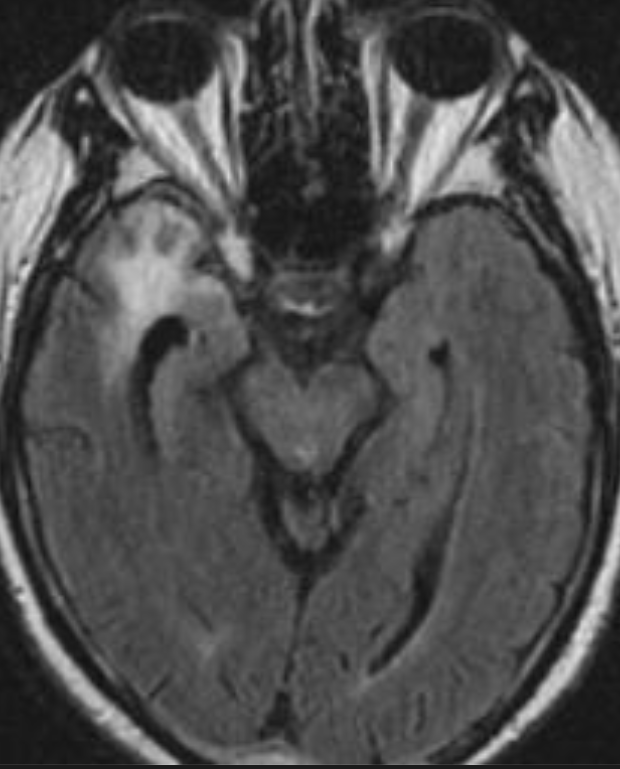
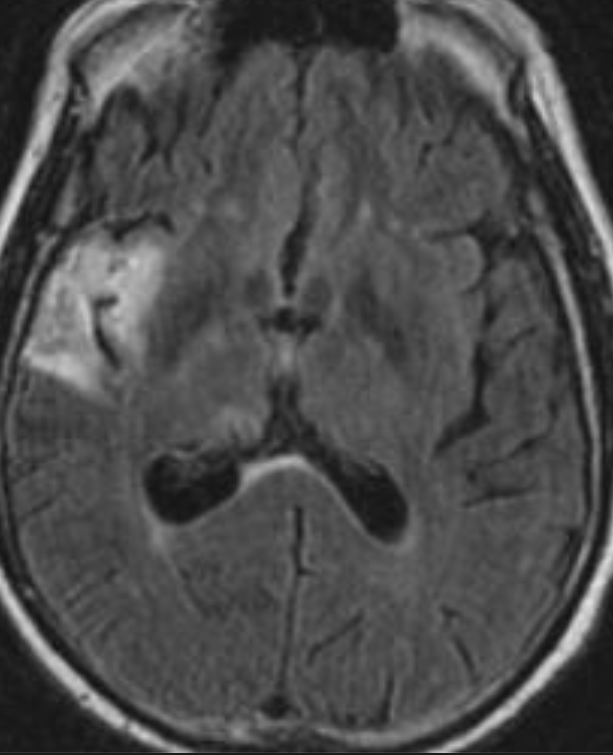
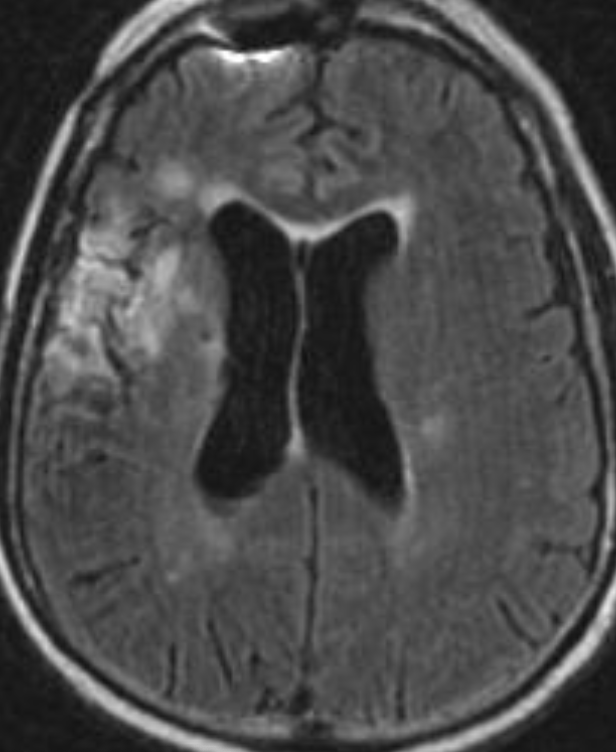
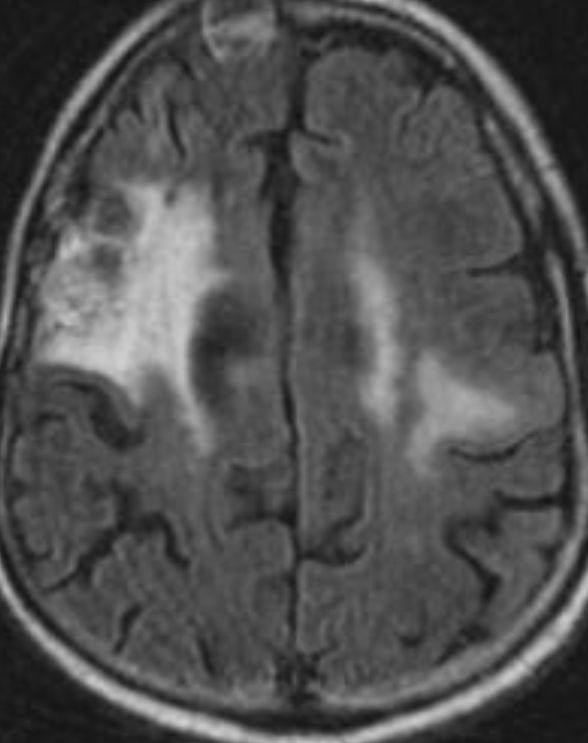
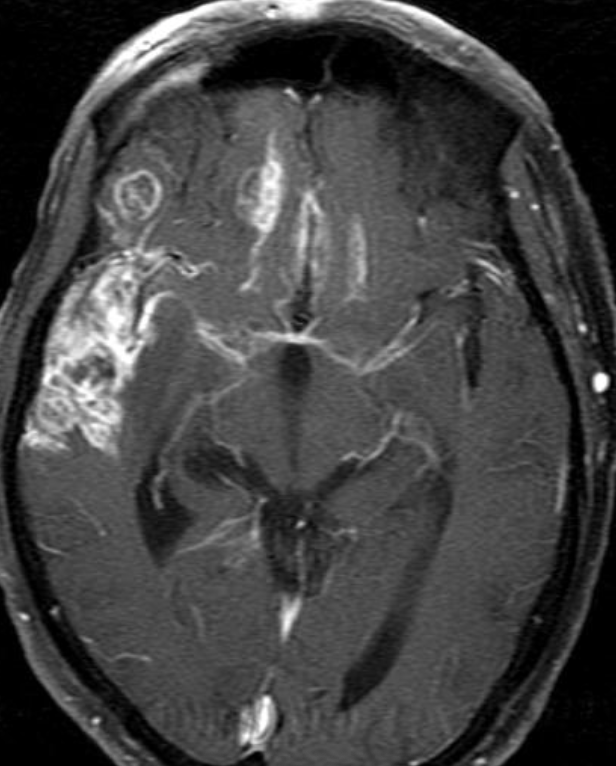
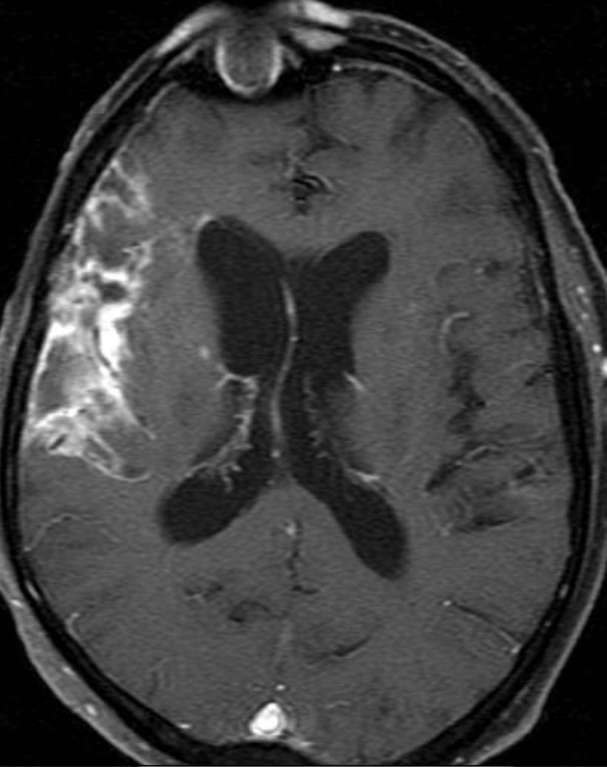
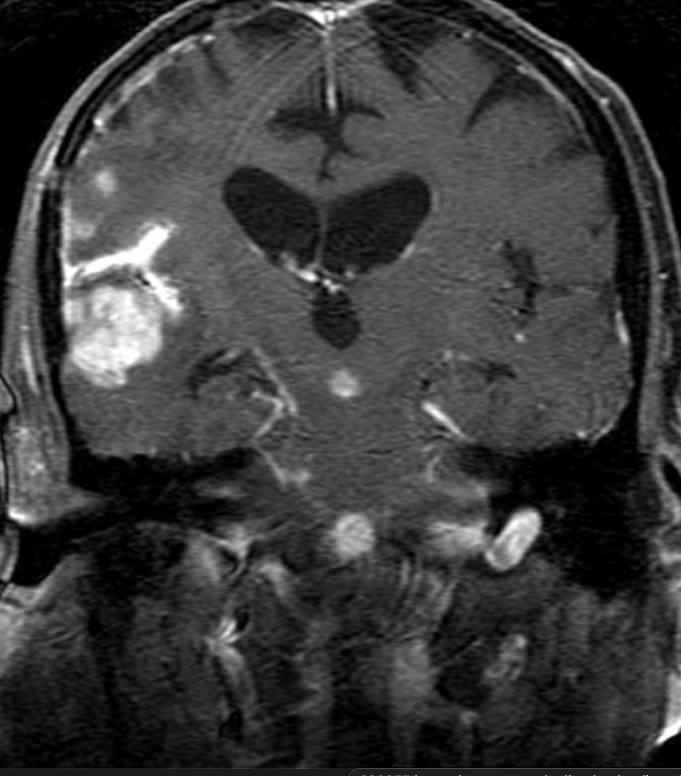
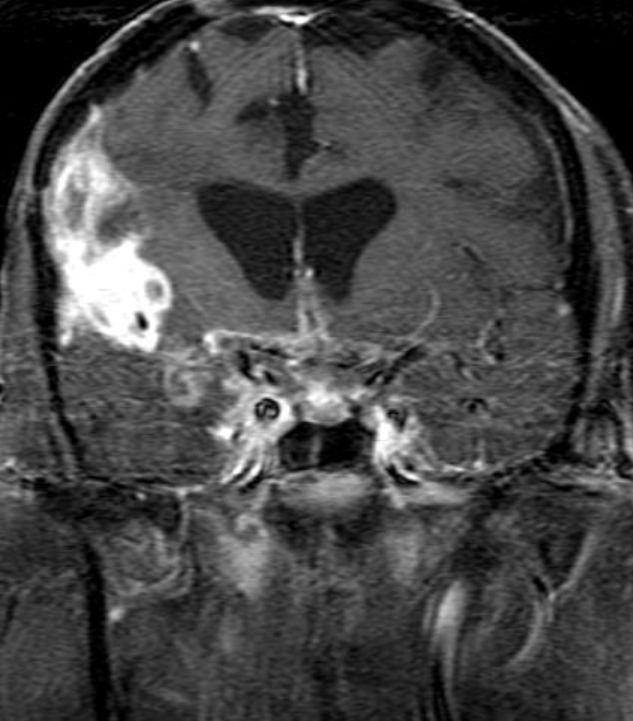
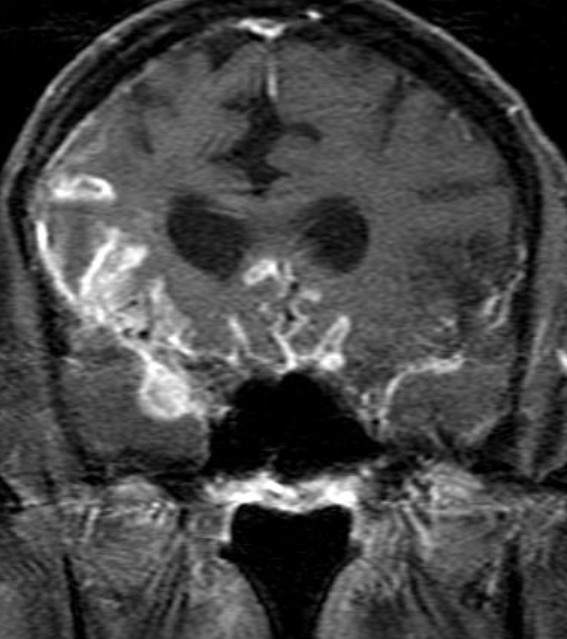
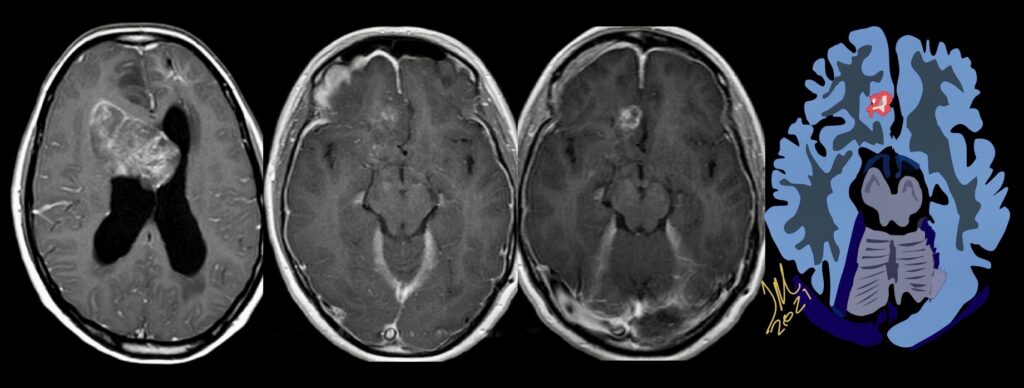
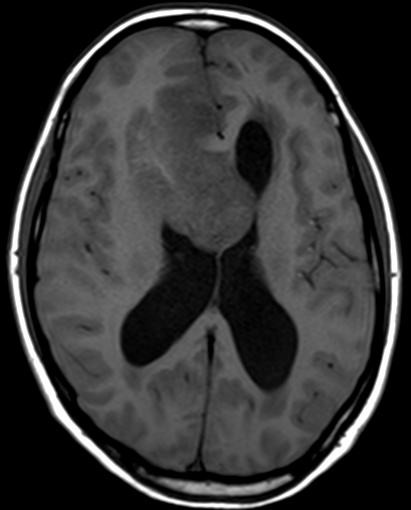
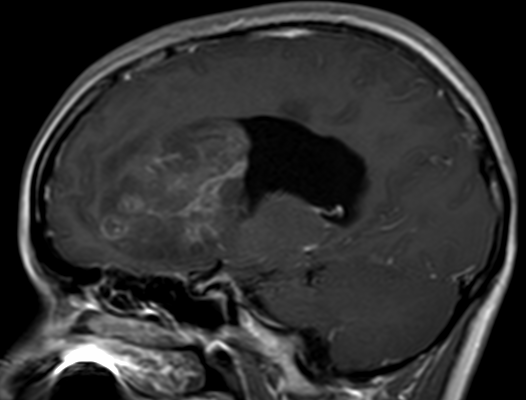
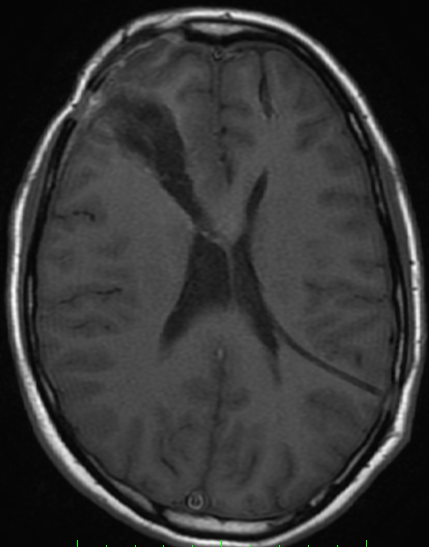
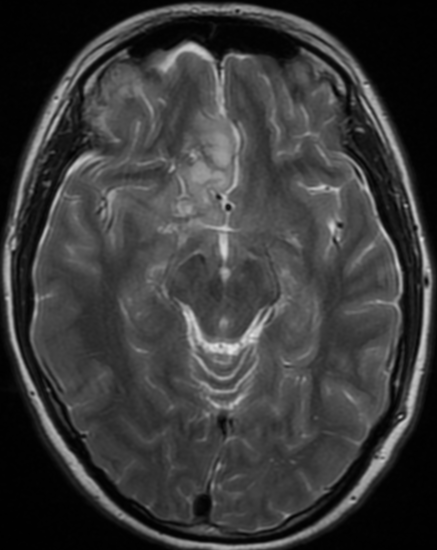
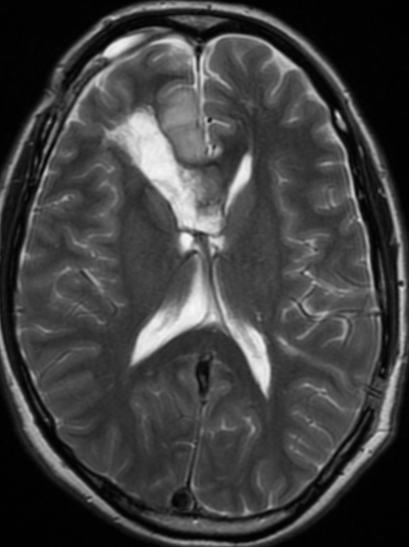
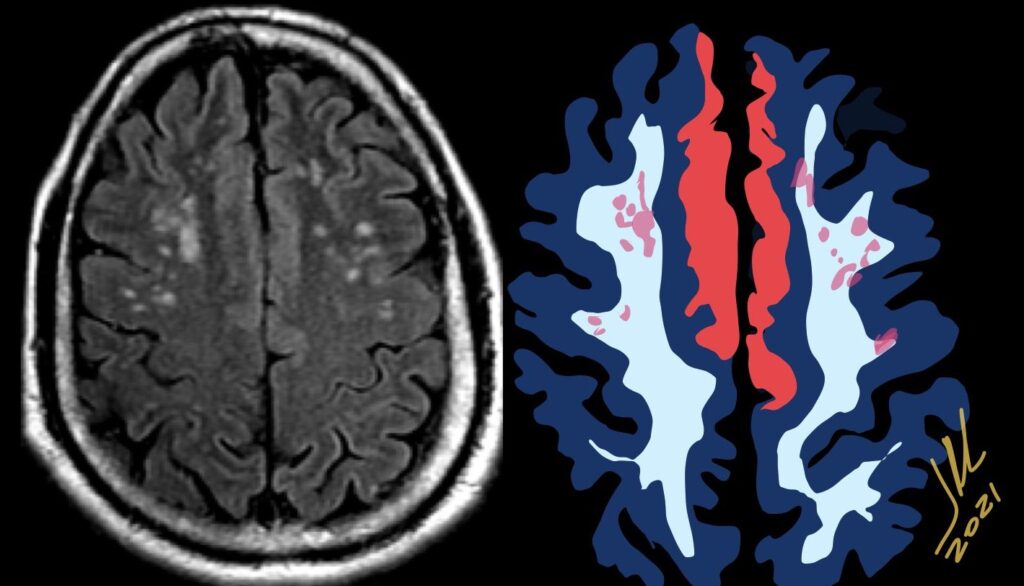
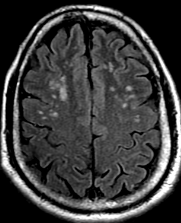
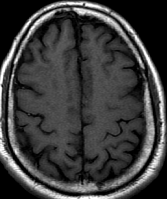
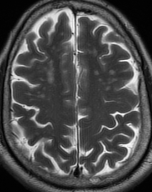
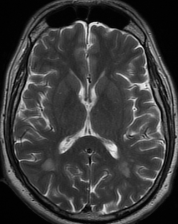
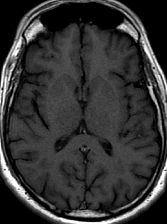
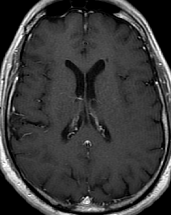
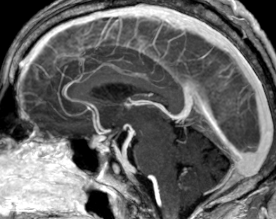
Cerebral radiation necrosis is a serious late delayed complication that manifests after a latency period of several months, although the range is broad and cases have been reported more than 10 years after irradiation. Histopathologic features of radiation necrosis include fibrinoid changes in the blood vessels, coagulative necrosis, demyelination, and gliosis. The disruption of the BBB (as visualized on gadolinium-enhanced images) may be mediated in part through vascular endothelial growth factor (VEGF) that is released in response to hypoxia. The reported incidence of radiation necrosis after RT for brain tumors ranges from 3% to 24%. Radiation necrosis is more likely to occur when high doses per fraction are administered, and combined use of chemotherapy with RT may play a role in development of treatment-related necrosis. Targeted therapy and immunotherapy may also increase the risk of treatment-related necrosis. Radiation necrosis typically develops at or adjacent to the original site of the tumor, the location that received the highest radiation dose. Radiation necrosis can also develop in part of the normal brain parenchyma that was included in the treatment field of a tumor outside the brain. In many cases, radiation necrosis is a self-limiting process that can be managed conservatively without intervention, although some patients need steroids for symptomatic relief.
Conventional MRI typically demonstrates an enhancing mass lesion with central necrosis and reactive edema. The enhancement pattern is often described as “spreading wavefront,” meaning that the margins of the enhancement are ill defined or “soap bubble–like” or “Swiss cheese–like,” meaning that the enhancing lesion includes central nonenhancing necrotic components of varying size. Multiple lesions are possible, and some lesions may develop distant from the original site of the tumor if the distant site was in the radiation field
Radiation necrosis.
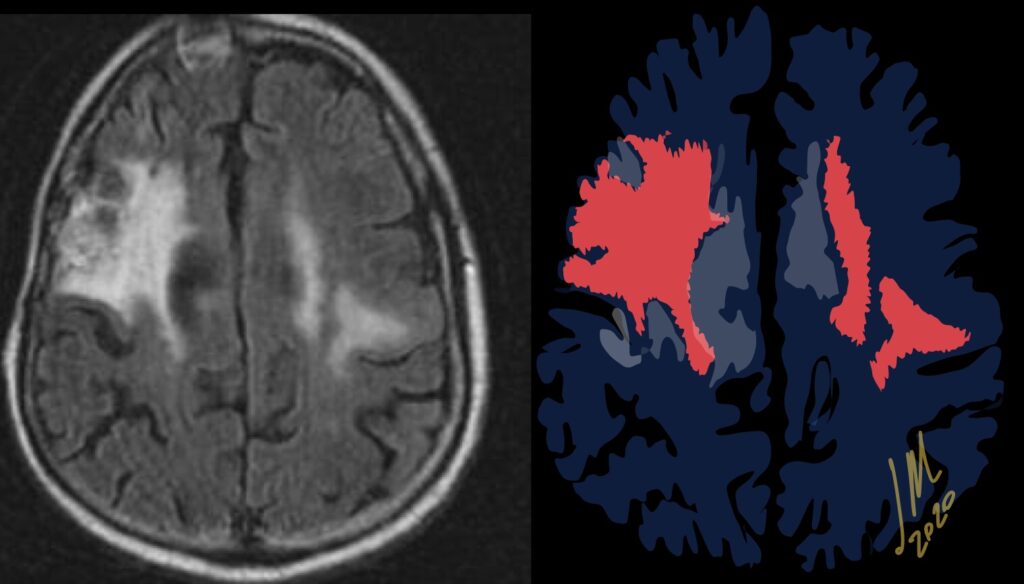
As radiation necrosis progresses with tumorlike growth, it can lead to marked shrinkage of the white matter and cortex and result in focal brain atrophy. The periventricular white matter is among the areas most susceptible to radiation necrosis. This may be explained by this neuroanatomic region having relatively poor blood supply from long medullary arteries that lack collateral vessels, making it vulnerable to ischemic effects produced by postirradiation vasculopathy. Imaging findings of radiation necrosis are not always irreversible and progressive but can be static, decrease, or even resolve at follow-up . White matter lesions were the earliest and most common manifestation after RT, followed by enhancing lesions, which often became necrotic with increasing size. Both white matter lesions and enhancing lesions were more likely than cysts to regress, and both could show complete resolution. Cysts were the least frequent pattern of radiation-induced injury and developed from enhancing lesions that exhibited necrosis
Conventional Imaging for Distinguishing Radiation Necrosis from Tumor Recurrence
Radiologic features of radiation necrosis at conventional imaging overlap with those of recurrent tumors, including high-grade primary brain tumors and brain metastases; therefore, image interpretation can be challenging. Biopsy of the suspicious lesion may be required for a definitive diagnosis, particularly in patients who are symptomatic and have worsening imaging findings over time. However, it is known that even at histopathologic analysis, residual or recurrent tumor mixed with radiation necrosis is a common finding.
Although not always reliable, some clinical or imaging features have been suggested in previous reports and may aid in diagnosis of radiation necrosis and tumor recurrence. First is the time elapsed since RT. Radiation necrosis usually manifests after a latency period of many months (12 months average) a new or worsening abnormality starting 3 years after RT is unlikely to be due to pure radiation necrosis. Corpus callosum involvement in conjunction with multiple enhanced lesions—with or without crossing of the midline and subependymal spread—were statistically significant, favoring predominant glioma recurrence
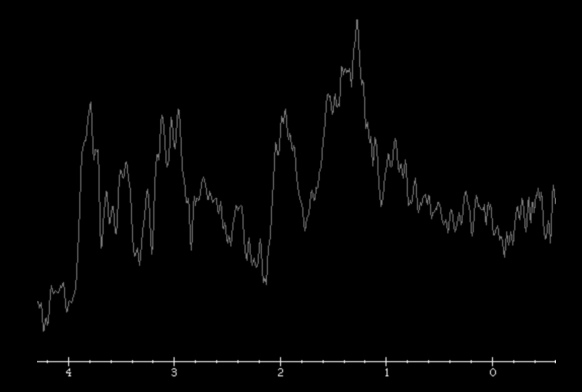
MR spectroscopy attempts to identify tumor recurrence by providing information on metabolic changes. Recurrent brain tumors exhibit high ratios of choline/creatine (Cho/Cr) and choline/N-acetylaspartate (Cho/NAA) whereas radiation necrosis exhibits increased lactate and lipid peaks .
Dynamic susceptibility contrast (DSC) MRI is the most widely used perfusion technique for brain tumors. DSC perfusion MRI relies on the T2- and T2*-shortening effects of gadolinium-based contrast agents and involves rapid imaging to capture the signal intensity changes due to the first passage of an intravenously administered contrast agent bolus.
The main parameter derived from DSC MRI in the context of brain tumors is relative cerebral blood volume (rCBV). rCBV is an indirect index of cerebral blood volume, generally calculated relative to region of interest (ROI) values in the contralateral normal white matter. Radiation necrosis typically causes hypoperfusion with reduced rCBV, whereas high-grade tumor recurrence results in high rCBV.
Oligo RT
The only method of distinguishing pseudoprogression and true tumor progression is to perform follow-up examinations of the patient because conventional MRI does not allow differentiation of the two conditions. Imaging may be regularly performed at 2–3-month intervals throughout the follow-up period although the frequency of imaging can be variable across institutions. In clinical practice, the following features can be helpful: (a) presence of symptoms and (b) methylation status of the MGMT gene promoter.
Recurrence
In the central nervous system, the effects of radiation can be roughly divided into effects on vascular endothelial cells and direct effects on neuroglial cells, in particular the oligodendroglial cells.
Vascular endothelial damage causes altered permeability, leading to vasogenic edema and disruption of the blood-brain barrier (BBB) or blood–spinal cord barrier. Preclinical studies suggest that endothelial damage may occur within the first 24 hours after irradiation. Endothelial damage can lead to other late vascular effects, such as telangiectasia, thrombosis, occlusion of small vessels, fibrinoid deposits, and hyaline thickening of vessel walls. As a result, ischemic stroke or hemorrhage may occur months to years after RT.
Oligodendrocytes are the most radiosensitive type of glial cell, with cell death occurring rather early after relatively low doses of irradiation. Radiation not only reduces the number of mature oligodendrocytes but also induces loss of their precursors, the oligodendrocyte type 2 astrocyte. This results in failure to replace normally turned over oligodendrocytes, with the eventual consequence being demyelination. In addition, there are changes in cellular composition such as increased numbers of reactive astrocytes (gliosis) and microglia. These reactive cells have been reported to produce reactive oxygen species, proinflammatory cytokines, and growth factors (eg, vascular endothelial growth factor [VEGF]), leading to a cycle of further cellular toxic effects and tissue damage .
Radiation-induced injury can be divided into three phases: acute, early delayed and late delayed. Acute and early delayed injuries are usually transient and reversible, whereas late delayed injuries are generally irreversible. Keys to recognizing radiation-induced changes at follow-up imaging are knowledge of (a) the amount of time elapsed since RT, (b) the location of the target lesion, and (c) the amount of normal structures included.
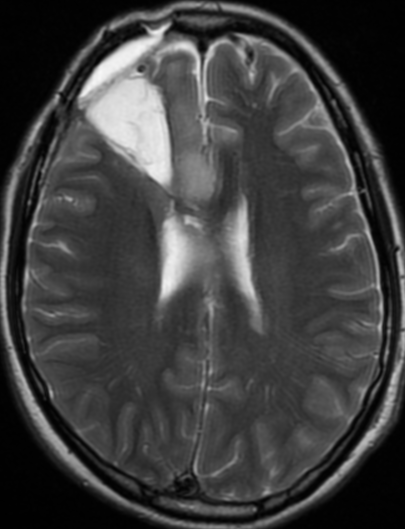
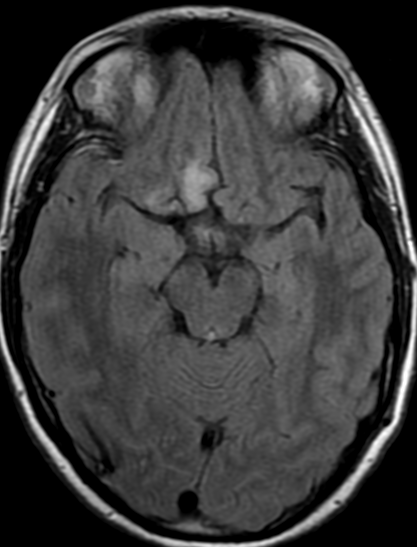
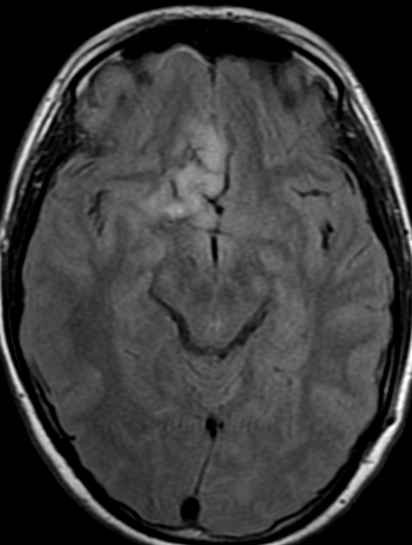
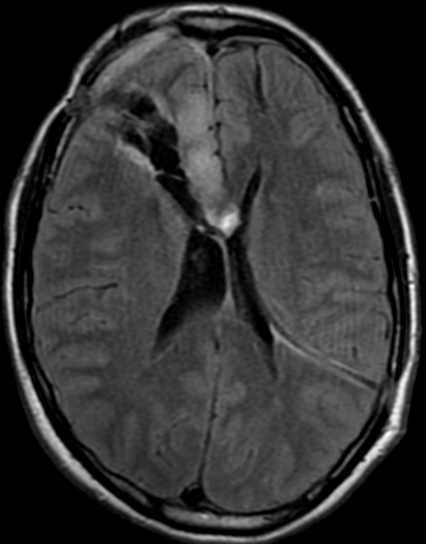
34F. First MRI. 2 months after surgery and 5 months later
Pseudoprogression is a subacute treatment-related effect with MRI features mimicking those of tumor progression. Patients can present with an increase in contrast enhancement and peritumoral edema at MRI. The diagnosis of pseudoprogression is typically made retrospectively on the basis of spontaneous improvement or stabilization of imaging findings without intervention.
Pseudoprogression typically develops in the setting of combined RT and temozolomide therapy for high-grade or low-grade glioma. but is also observed with immune checkpoint inhibitors in combination with RT for brain metastases and in cases where chemotherapy-infused wafers were placed in the surgical cavity. Pseudoprogression usually develops within 3 months after completion of chemoradiation therapy and is often clinically asymptomatic. The timing of pseudoprogression is earlier than the typical period in which radiation necrosis is described after RT alone; therefore, it is often classified as an early delayed reaction to radiation. Pseudoprogression is most likely induced by a marked local tissue reaction with an inflammatory component, edema, and abnormal vessel permeability causing new or increased enhancement at MRI. Pathologically, pseudoprogression is found to correspond to gliosis and reactive radiation-induced changes without evidence of viable tumor
first control 2 months after surgery
The only method of distinguishing pseudoprogression and true tumor progression is to perform follow-up examinations of the patient because conventional MRI does not allow differentiation of the two conditions. Imaging may be regularly performed at 2–3-month intervals throughout the follow-up period although the frequency of imaging can be variable across institutions.
In clinical practice, the following features can be helpful: (a) presence of symptoms and (b) methylation status of the MGMT gene promoter. REF;Nov 20 2020 https://doi.org/10.1148/rg.2021200064
5 months follow up pseudoprogression, Treatment with Rt and temozolomide
The current standard treatment protocol for glioblastomas is surgical resection followed by 6 weeks of radiation therapy plus concomitant temozolomide chemotherapy (CCRT) and 6 cycles of adjuvant temozolomide chemotherapy. This protocol increases median survival from 12 to 15 months. Tumors with hypermethylation of the O6-methylguanine-DNA methyltransferase promoter gene show pseudoprogression more frequently. Enlarged enhancing lesions on conventional MR images may actually represent pseudoprogression in up to 46.8%–64% of cases.
The Response Assessment in Neuro-Oncology (RANO) Working Group proposed that within the first 12 weeks of completion of radiation therapy, when pseudoprogression is most prevalent, tumor progression can only be determined if most of the new enhancement is outside the radiation field or if there is pathologic confirmation of progressive disease.
MR imaging techniques such as DWI and dynamic susceptibility contrast PWI. On DWI, ADC values are higher in necrotic tissue than in recurrent tumor tissue because of the high cellularity of tumor tissue. However, the use of DWI is limited due to the heterogeneity of tumor content. Reduced diffusion represents not only highly cellular tumor areas but also inflammatory processes.
On PWI, high relative cerebral blood volume (rCBV) is considered active neovascularization and viable tumor. rCBV > 1.47 had 81.5% sensitivity and 77.8% specificity for differentiating pseudoprogression from tumor progression. However, rCBV analysis has limitations because most lesions have variable tumor fractions; therefore, mean rCBV and histogram-based metrics may be influenced by the rCBV from both tumoral and nontumoral components. Reference : https://doi.org/10.3174/ajnr.A3876
the appearance of enhancing lesions on MR imaging within the first 6 months after completion of chemoradiation therapy poses a challenge because it can reflect true progression (TP) or treatment-related changes known as pseudoprogression (PsP). PsP occurs in approximately a third of all patients with glioblastoma. Accurate identification of PsP and TP is critical because patients with TP may require a change in therapeutic strategy while those with PsP may not. The heterogeneity and variability in response did not allow differentiating TP from PsP simply by visual inspection of the parametric maps. However, a quantitative analysis of DTI parameters and rCBVmax from the enhancing regions of the lesion demonstrated better assessment of treatment response in patients with glioblastomas.
Identification of TP
Early identification of TP could prevent further delays in repeat surgery or enrollment in alternative clinical trials. The LRM analysis indicated that the best model to distinguish TP from PsP or mixed responses was based on FA, CL, and rCBVmax. Higher anisotropy values have been reported in glioblastomas compared with brain metastases and primary cerebral lymphomas. High FA in glioblastomas is probably related to the orientation of overproduced extracellular matrix.
Identification of PsP
Accurate identification of PsP is critical for patient management because unnecessary repeat surgery/biopsy can be avoided in these patients and they can continue on an effective temozolomide regimen with standard imaging follow-up of 3–6 months, thereby reducing patient care costs. Logistic regression analysis showed that the best model to differentiate PsP from TP and mixed response included FA and rCBVmax.
Pseudoprogression is predominantly a subacute treatment-related reaction. Pathologically, it corresponds to gliosis and radiation-induced reactive changes including disruption of the BBB, inflammation, increased permeability, and edema. These changes cause increased enhancement on MR imaging and can mimic TP. combination of FA and rCBVmax can help in identifying PsP from TP or mixed response.
Identification of Mixed Response
On a practical level, posttreatment new enhancing lesions usually contain a mixture of viable neoplasm and treatment-induced changes, and a more accurate assessment of the relative contribution of each entity can guide clinical decision-making. However, most previous studies have attempted to only differentiate between PsP and TP. REF; American Journal of Neuroradiology January 2016, 37 (1) 28-36; DOI: https://doi.org/10.3174/ajnr.A4474
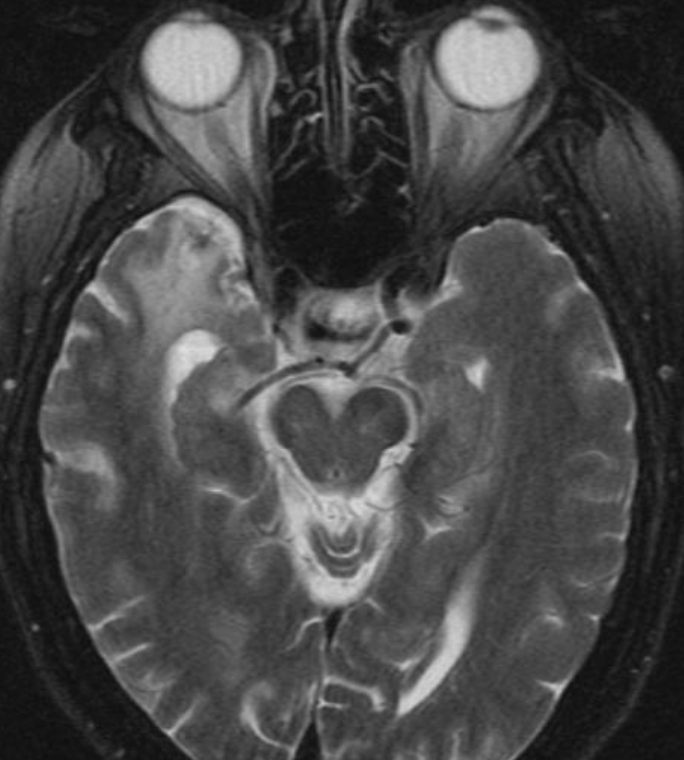
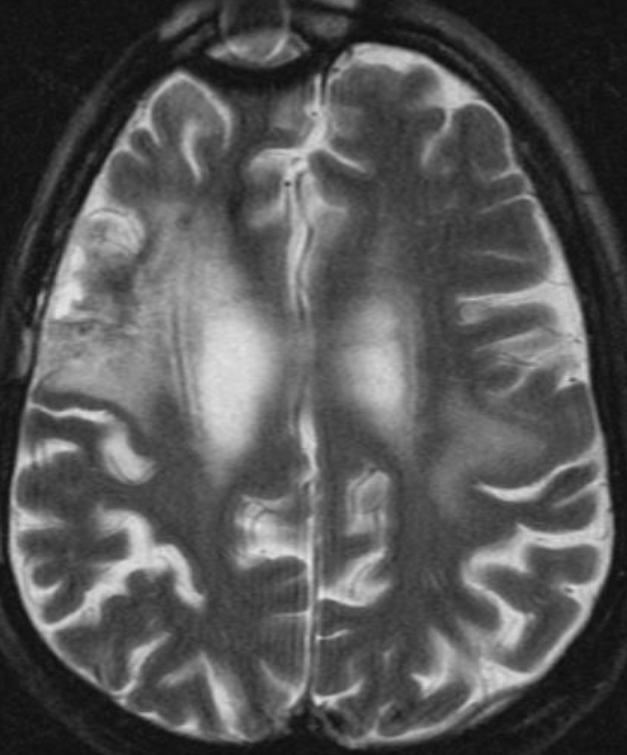
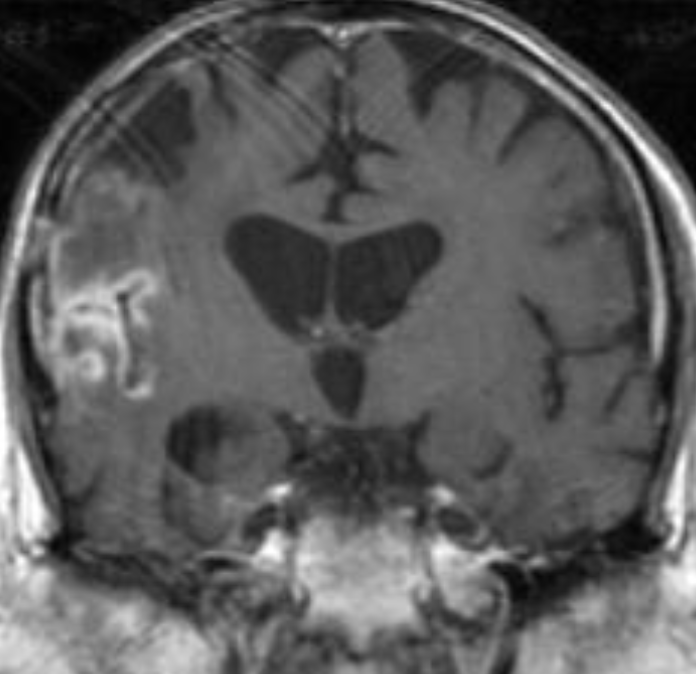
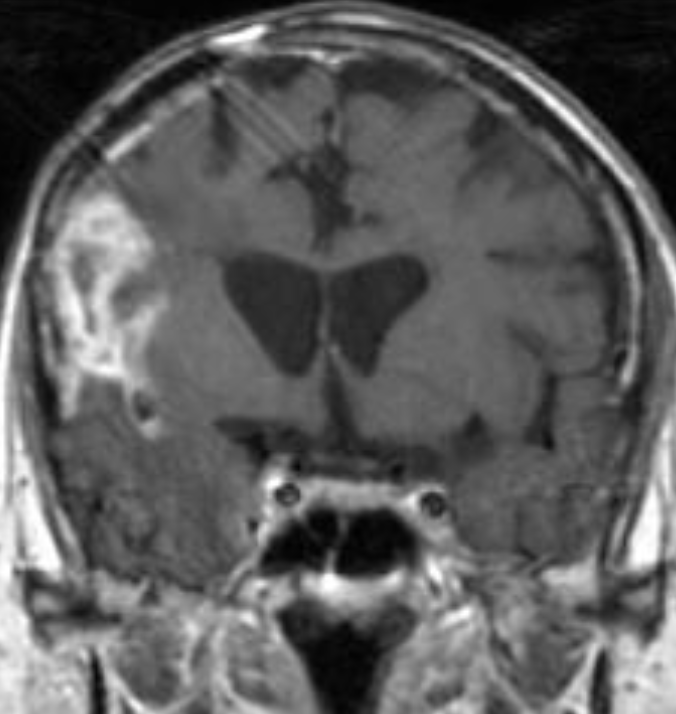
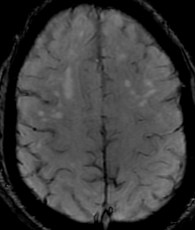
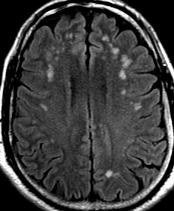
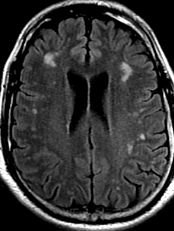
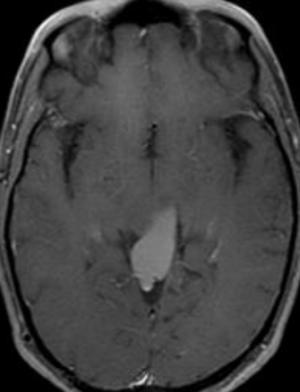
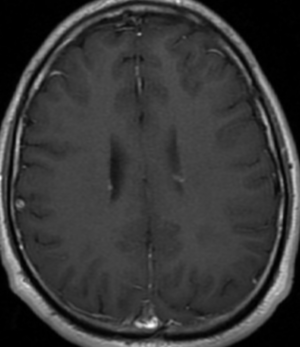
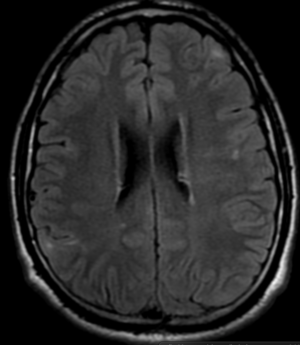
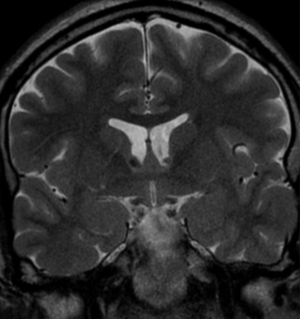
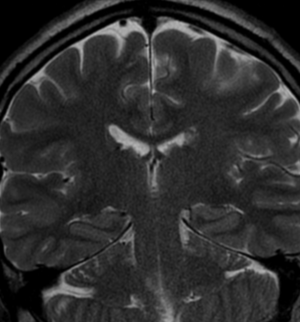
50M. patient who had covid course 70 days before without any respiratory manifestation only with persistent brain fog.
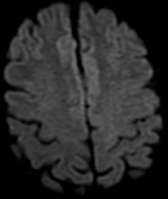
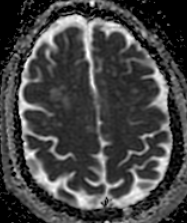
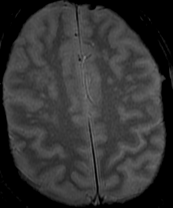
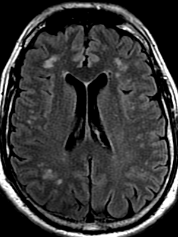
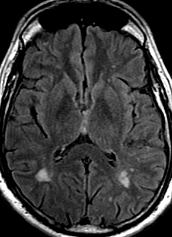
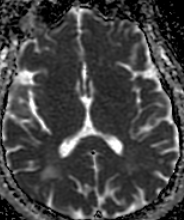
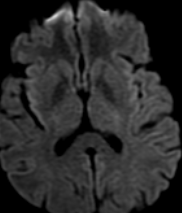
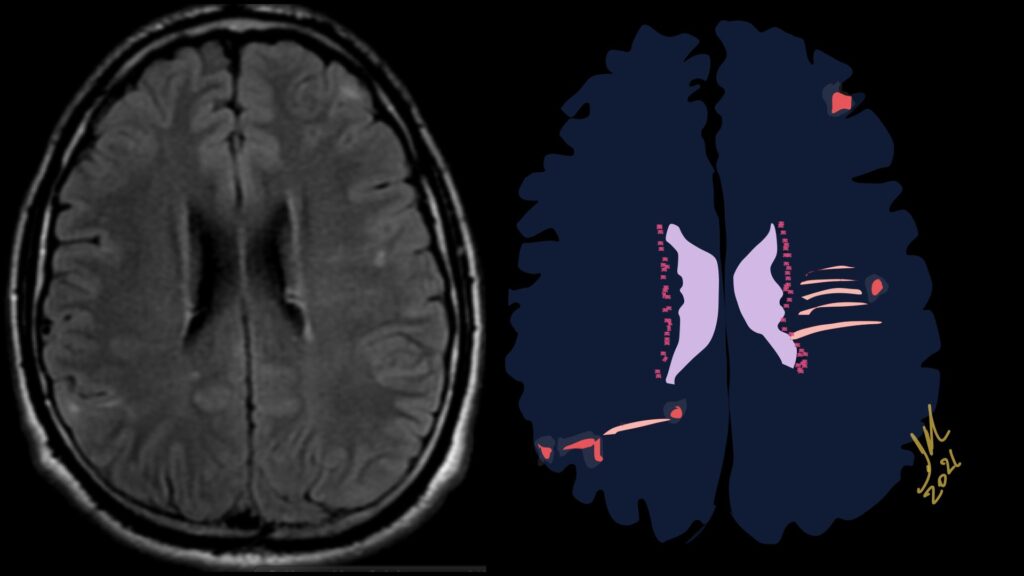
The most frequent diagnoses made at brain MR imaging in patients with COVID-19 are acute and subacute infarcts. Other common findings included a constellation of leukoencephalopathy and microhemorrhages, leptomeningeal contrast enhancement, and cortical FLAIR signal abnormality.
Meningitis and encephalitis are uncommon in patients with COVID-19 and neurologic symptom. Also cortical FLAIR hyperintensity should be secondary to encephalitis. Nonspecific cortical pattern of T2 FLAIR hyperintense signal with associated restriction diffusion that may be caused by systemic toxemia, viremia and-or hypoxic effects.
Cortical FLAIR signal abnormality has a broad differential diagnosis and can include encephalitis, postictal state, PRES, as well as acute ischemia.
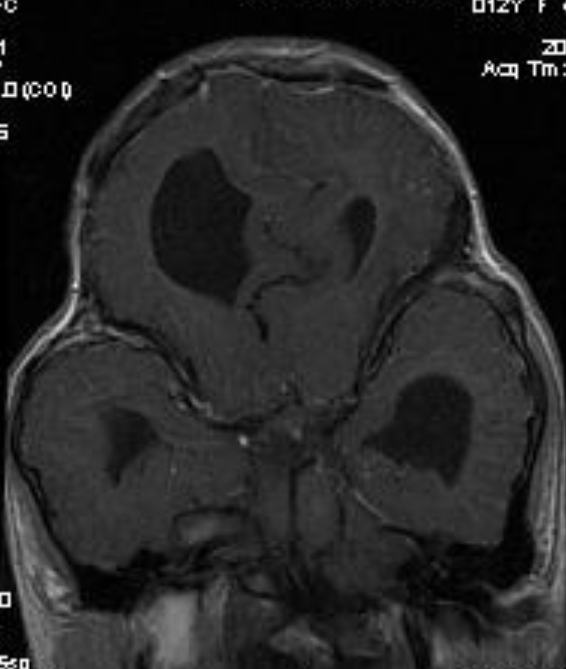
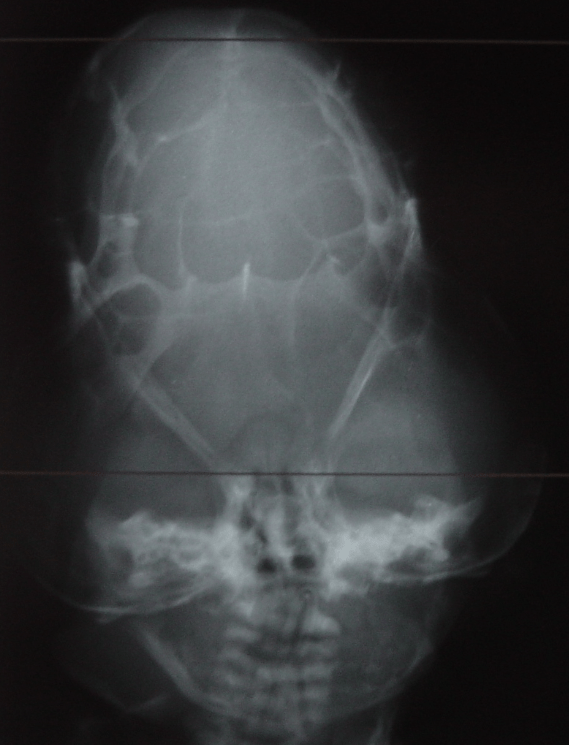
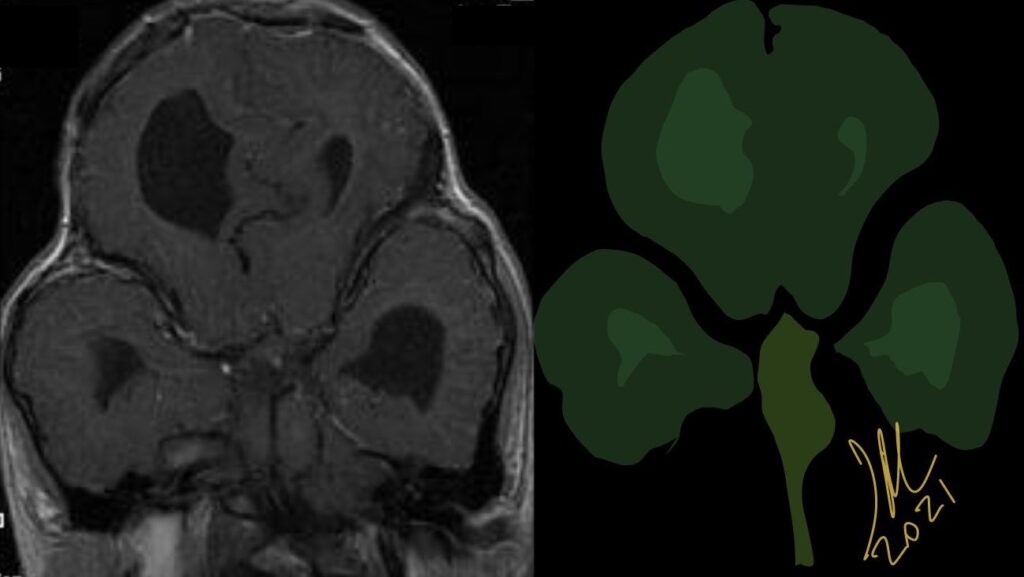
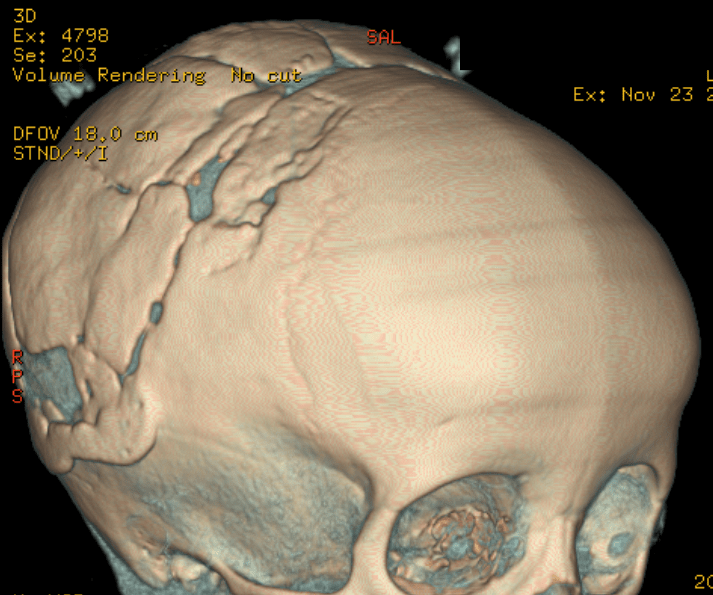
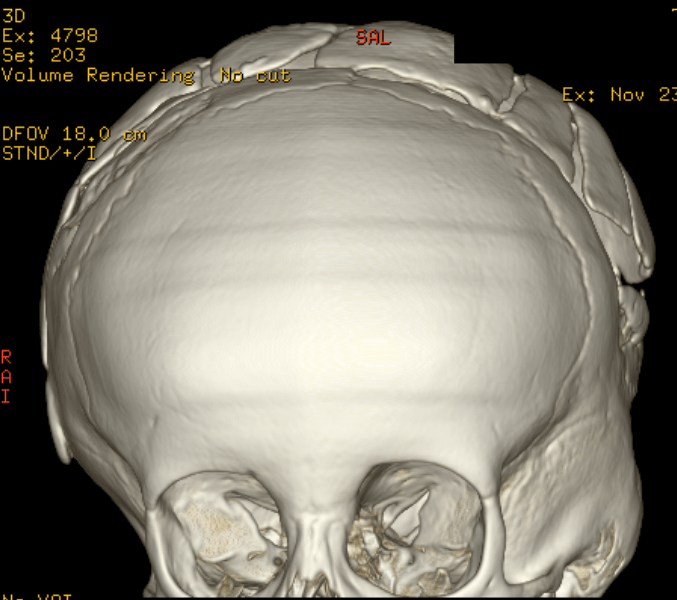
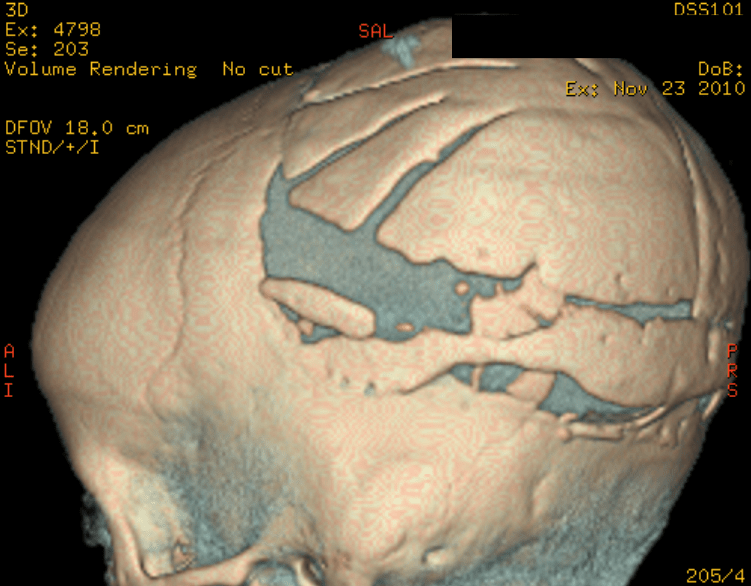
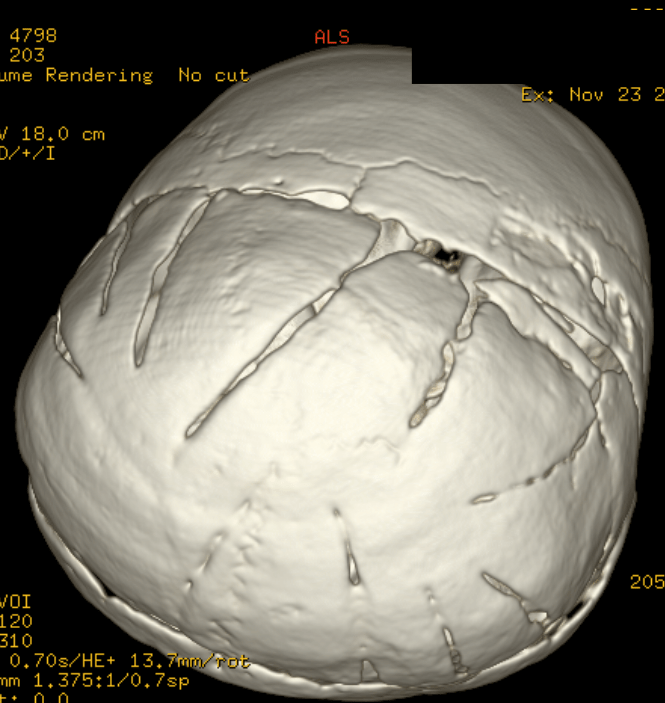
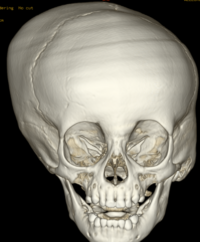
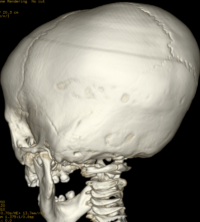
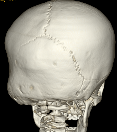
Cloverleaf skull syndrome is an abnormal configuration of the calvaria classified as craniosynostosis, consisting of premature ossification of cranial sutures. It is a deformity characterized by a remarkable enlargement of the head, with a trilobed configuration of the frontal view, resembling a three leaved clover. This abnormality occurs as a result from a severe alteration in the development of the skull, with premature synostosis of some cranial sutures, most commonly the coronal and lambdoid sutures, in association with hydrocephalus, leading to a marked bulging of the head in the region of the anterior fontanel and laterally in the temporal regions, with the typical appearance of a «cloverleaf». Syndromic and nonsyndromic presentations have been reported. Because of the anomalies both in the calvaria and in the skull base and face, this is one of the craniosynostosis currently requiring the most complex multidisciplinary approach.
The precise etiopathogenesis of this syndrome is still to be completely known, with theories involving altered membranous-osseous and/or endochondral ossification, generalized chondrodysplastic process, and a possible vascular origin associated with the abnormal osteoclastic resorption. Recently, genetic investigations have contributed to advances in the understanding of the molecular basis of some craniosynostosis syndromes, highlighting mutations in the genes FGFR1, FGFR2, FGFR3, TWIST and MSX2.
The diagnosis of such a syndrome can be made in the prenatal period by means of ultrasonography, which detects the altered cranial morphology and hydrocephalus. Traditionally, the diagnosis occurs during routine prenatal follow up at the second gestational trimester. However, with the increasing use of obstetric ultrasonography at the first gestational trimester, such alterations may be detected increasingly earlier over the gestation.
Pfeiffer syndrome is a form of pansynostosis of the cranial sutures with associated limb abnormalities. There are 3 types of Pfeiffer syndrome with type I (classic) being the mildest form of the disorder. Midface hypoplasia and abnormal skull shape are minimal in this form of the disease, and intelligence is more likely to be normal in these patients. Type II is more severe and results in the formation of a “cloverleaf” skull (our case), also known as kleeblattschädel, and the degree of cranial abnormality is more pronounced. Pfeiffer syndrome type II is commonly fatal in infancy due to its marked compression on the intracranial structures. Pfeiffer syndrome type III is also more severe than Pfeiffer syndrome type I; however, the cloverleaf skull phenotype is less common in Pfeiffer syndrome type III.
Pfeiffer syndrome type II is caused by a mutation in the gene for fibroblast growth factor receptor 2 (FGFR2) on the long arm of chromosome 10, a gene that encodes a cell membrane protein responsible for cell differentiation. Pfeiffer syndrome type I has an association with FGFR1, a different gene on the short arm of chromosome 8. Mutations in the FGFR genes are also implicated in Apert, Beare-Stevenson, Crouzon, Jackson-Weiss, and Muenke syndromes, all diseases that exhibit craniosynostosis and/or facial malformations to varying degrees. The pattern of inheritance for Pfeiffer syndrome is autosomal dominant with cases of Pfeiffer syndrome types II and III due primarily to sporadic de novo mutations.
Surgical correction of the deformities is complex. Midface hypoplasia and choanal atresia are common, as was the case in this patient. Typical steps involved in the reconstruction include ventriculostomy or shunt placement to relieve increased intracranial pressure, fronto-orbital advancement, and posterior skull remodeling
The cloverleaf skull deformity (kleeblattschädel) was first described by Holtermuller and Wiedemann. The upper and lower leaves of the cloverleaf are formed by a ring of bone that divides the upper and lower leaves of the cloverleaf, and there is a honeycomb pattern of the inner vault of the skull. Crowding of the posterior fossa and subsequent hindbrain herniation are common. Kleeblattschädel can be seen in severe forms of Crouzon and Apert syndromes, Saethre-Chotzen syndrome, Carpenter syndrome, and thanatophoric dysplasia, though Pfeiffer syndrome has been reported to account for approximately 15%-20% of cases. Intellectual impairment observed in patients with cloverleaf skull is most likely a result of the intracranial mass effect from the calvarial deformity rather than an intrinsic brain abnormality, as normal intelligence by 4 years of age has been reported in some cases of patients undergoing a staged reconstruction of kleeblattschädel
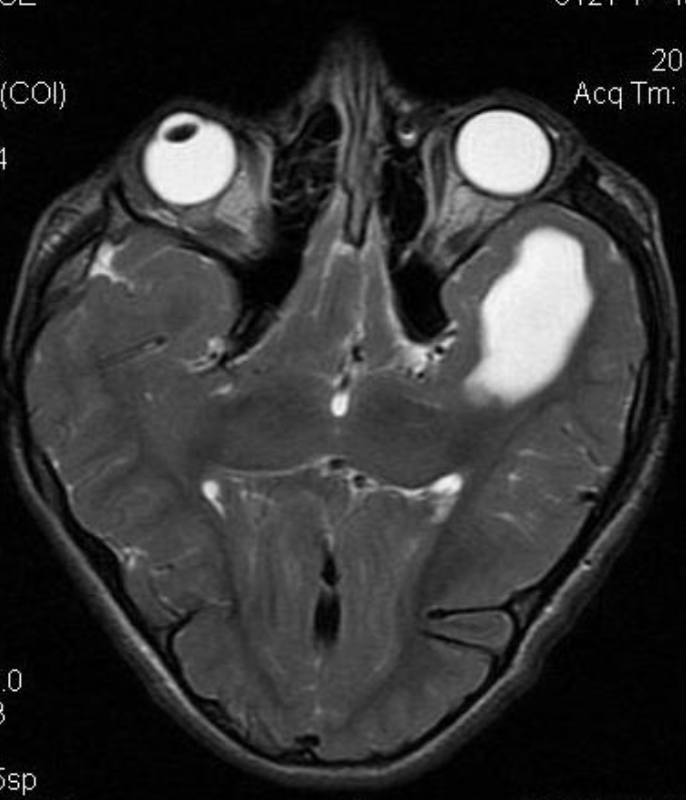
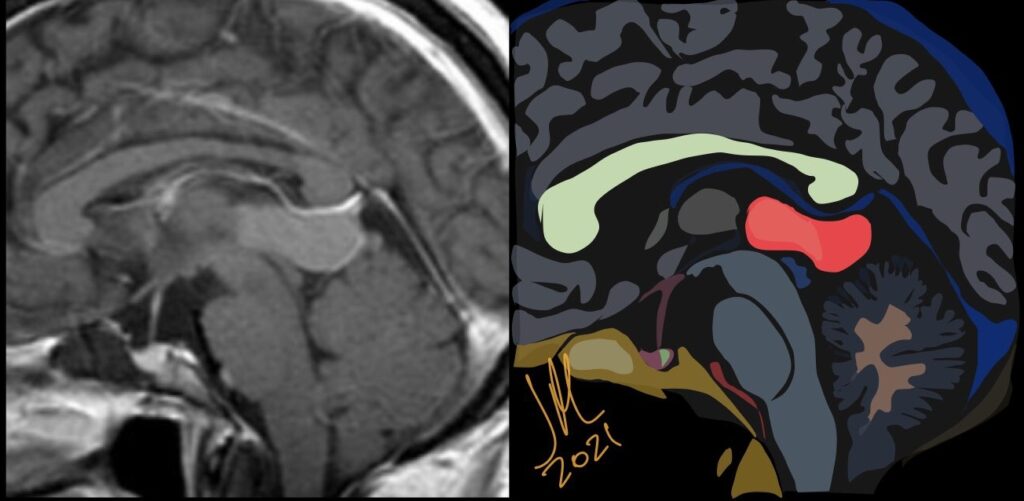
The pineal localization is a very rare form of this intracranial lesion. It represents 0,2-1% of all intracranial tumors. Cushing was the first to report the pineal localization of the epidermoid cyst in 1928. The clinical presentation is often characterized by parinaud’s syndrome and hydrocephalus. Hemiparesis and cerebellar signs can also be noticed
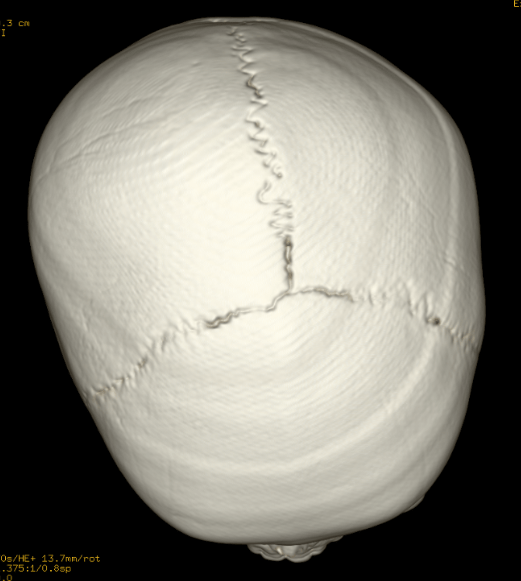
Craniosynostosis refers to the premature closure of the cranial sutures. The skull shape then undergoes characteristic changes depending on which sutures close early. 3D CT MIP can depict suture patency, extent of synostosis (ie, complete versus incomplete bone bridging), fracture extent and conspicuity, and three-dimensional calvarial deformity as a single set of projections in children with suspected craniosynostosis or skull fracture.
Types of craniosynostosis
There are several types of craniosynostosis. Most involve the fusion of a single cranial suture. Some complex forms of craniosynostosis involve the fusion of multiple sutures. Most cases of multiple suture craniosynostosis are linked to genetic syndromes and are called syndromic craniosynostosis.
The term given to each type of craniosynostosis depends on what sutures are affected. Types of craniosynostosis include:
Sagittal (scaphocephaly). Premature fusion of the sagittal suture that runs from the front to the back at the top of the skull forces the head to grow long and narrow. Sagittal craniosynostosis results in a head shape called scaphocephaly and is the most common type of craniosynostosis.
Coronal. Premature fusion of one of the coronal sutures (unicoronal) that run from each ear to the top of the skull may cause the forehead to flatten on the affected side and bulge on the unaffected side. It also leads to turning of the nose and a raised eye socket on the affected side. When both coronal sutures fuse prematurely (bicoronal), the head has a short and wide appearance, often with the forehead tilted forward.
Metopic. The metopic suture runs from the top of the bridge of the nose up through the midline of the forehead to the anterior fontanel and the sagittal suture. Premature fusion gives the forehead a triangular appearance and widens the back part of the head. This is also called trigonocephaly.
Lambdoid. Lambdoid synostosis is a rare type of craniosynostosis that involves the lambdoid suture, which runs along the back of the head. It may cause one side of your baby’s head to appear flat, one ear to be higher than the other ear and tilting of the top of the head to one side.
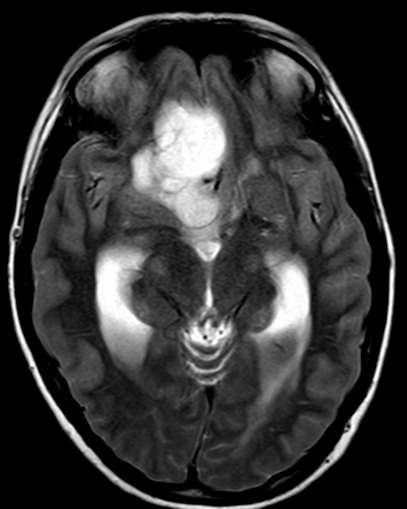
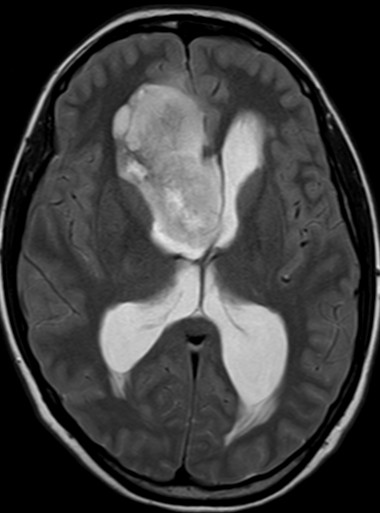
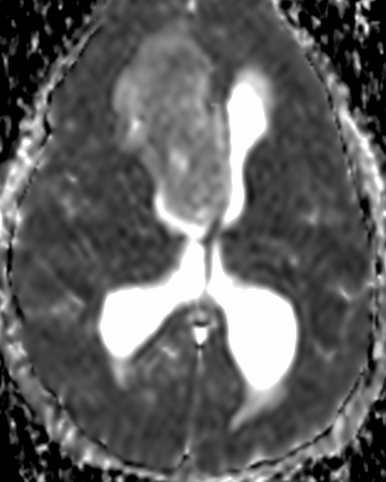
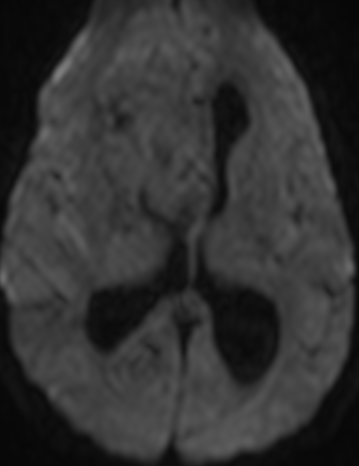
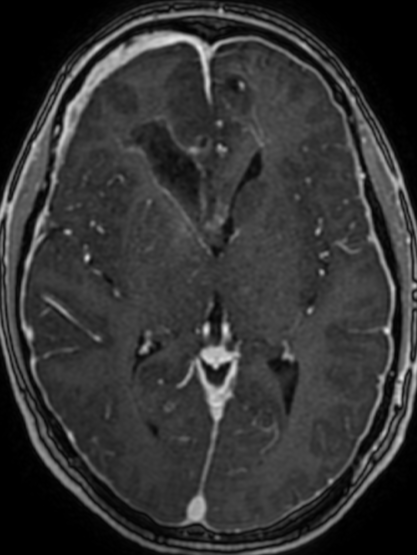
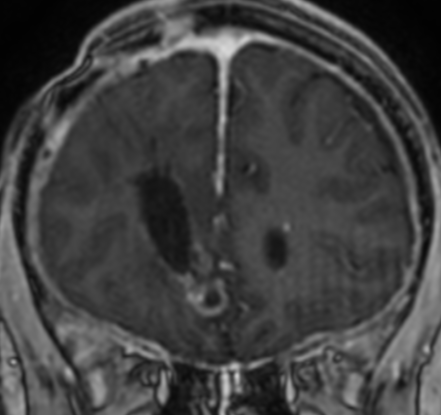
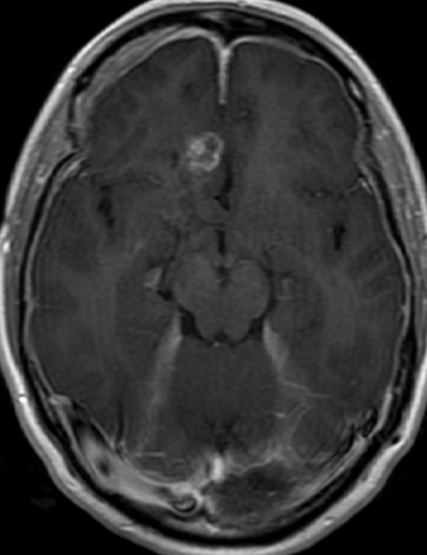
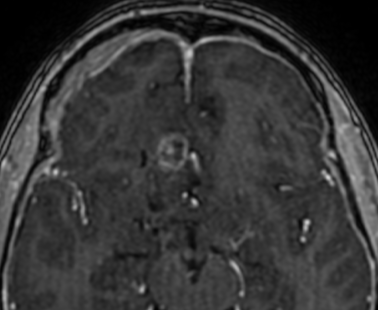
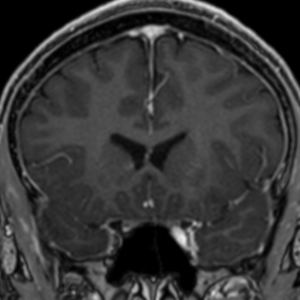
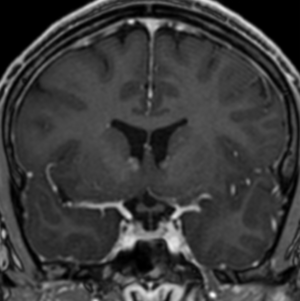
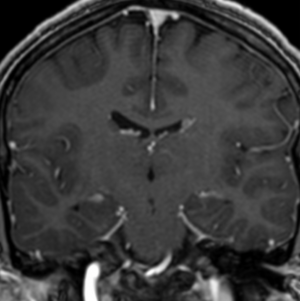
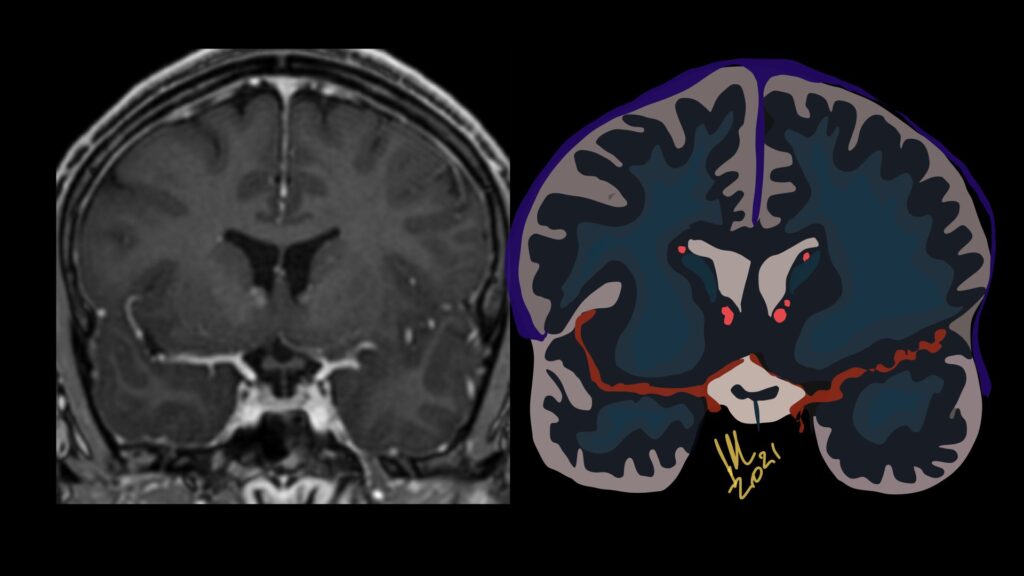
Imaging features, without any physical findings or symptoms, can also be used to make the diagnosis of TS. These unique diagnostic findings include the presence (on CT, MR, US) of multiple subependymal nodules (SENs; especially when calcified) or wedge-shaped cortical calcifications, intraventricular tumor consistent with subependymal giant cell astrocytoma (SGCA), and multiple cortical or subcortical “tubers” (especially if associated with subcortical white matter edema). In addition, typical linear abnormalities in the white matter may be found, Cystic lacune–like lesions have also been described in up to 44% of TS patients.
Radial Lines and subcortical tubers and subependymal nodules (SENs).
The SENs, although quite obvious on CT can be subtle on MR. The signal intensity patterns are variable and can change over time. Some SENs may have a “target” appearance on T2-weighted MRI, with a central hyperintensity reminiscent of the gyral core seen in the cortical tubers. the center of a SEN can be bright on the T1-weighted image.This appearance could be related to the paradoxical T1 shortening that occurs with dispersed microscopic calcifications. SENs show gadolinium enhancement, often clustered about the foramen of Monro. The SENs are histologically similar to the cortical hamartomas but are smaller and arise primarily in the striothalamate groove along the lateral margin of the lateral ventricles.
The SENs do not appear to grow; however, they do calcify progressively during the first two decades, so that by age 20 years virtually 100% are hyperdense, This presence of calcification allows SENs to be distinguished from the otherwise similar-appearing subependymal gray matter heterotopias. Heterotopic gray matter is isodense and isointense to normal gray matter, without contrast enhancement
Tuberous sclerosis complex (TSC) or Bourneville-Pringle syndrome is a Multisystem genetic disorder with epilepsy, multiorgan tumors, and hamartomas. Spectrum of central nervous system, hamartomas, all contain dysplastic neurons and giant (balloon) cells. Caused by mutation in TSC1 or TSC2 gene. Now considered infantile (developmental) tauopathy. Tau abnormally expressed in many of dysmorphic neurons and glial cells of TSC.