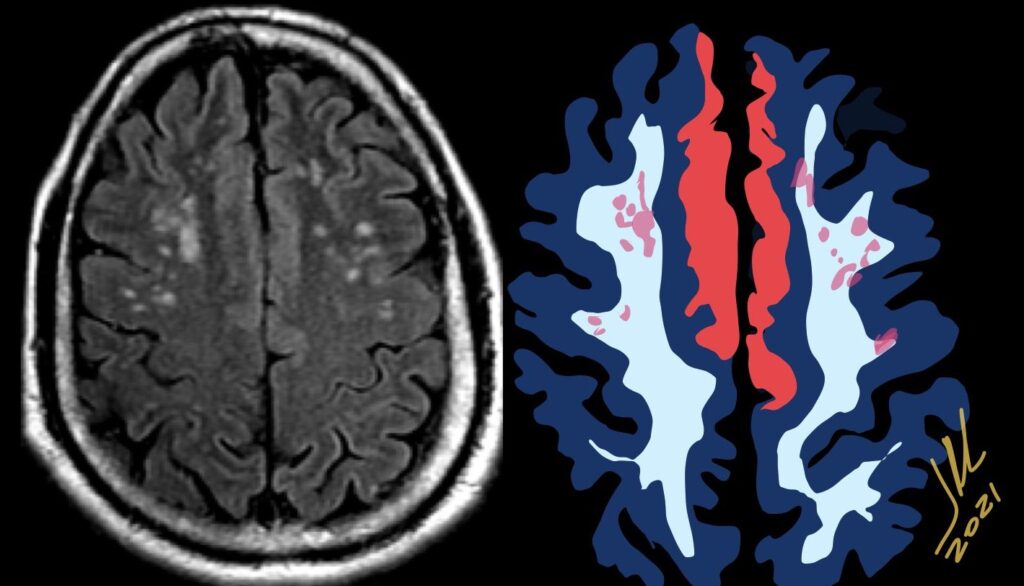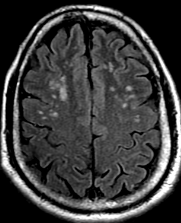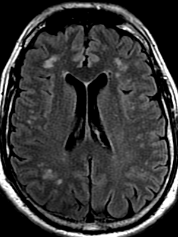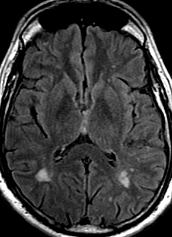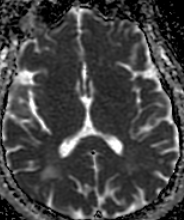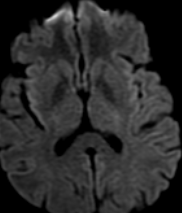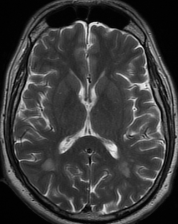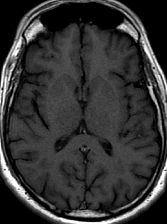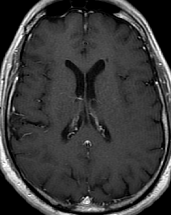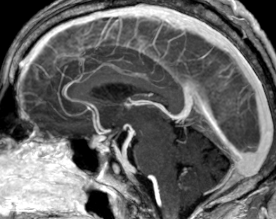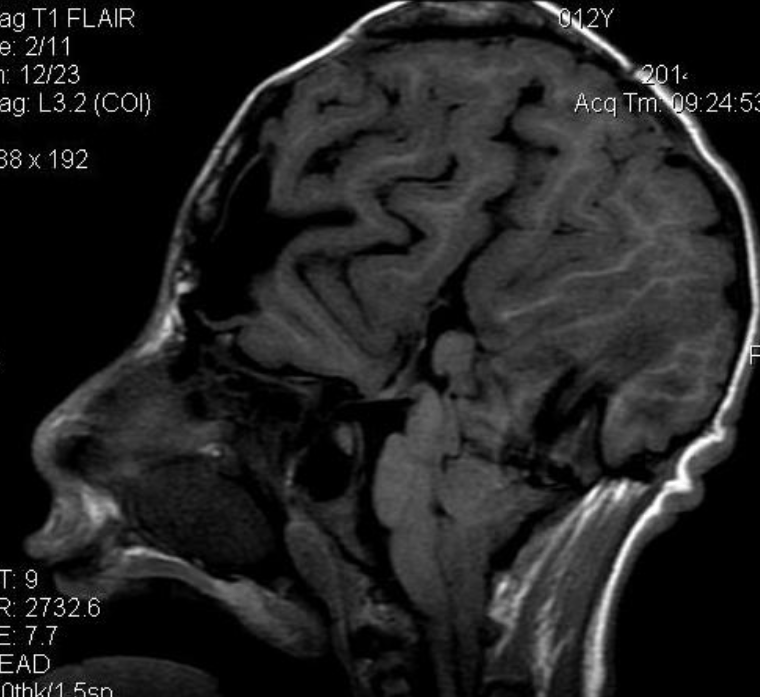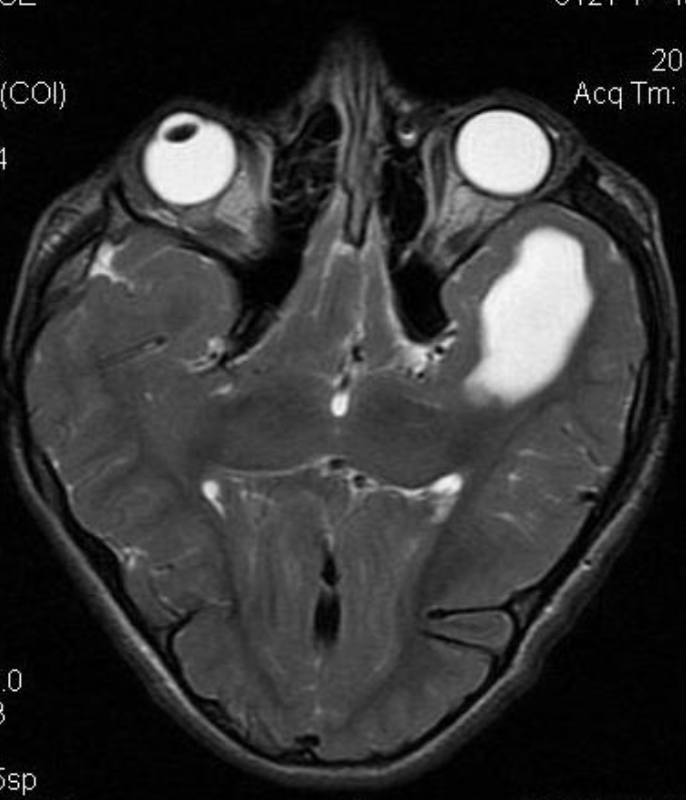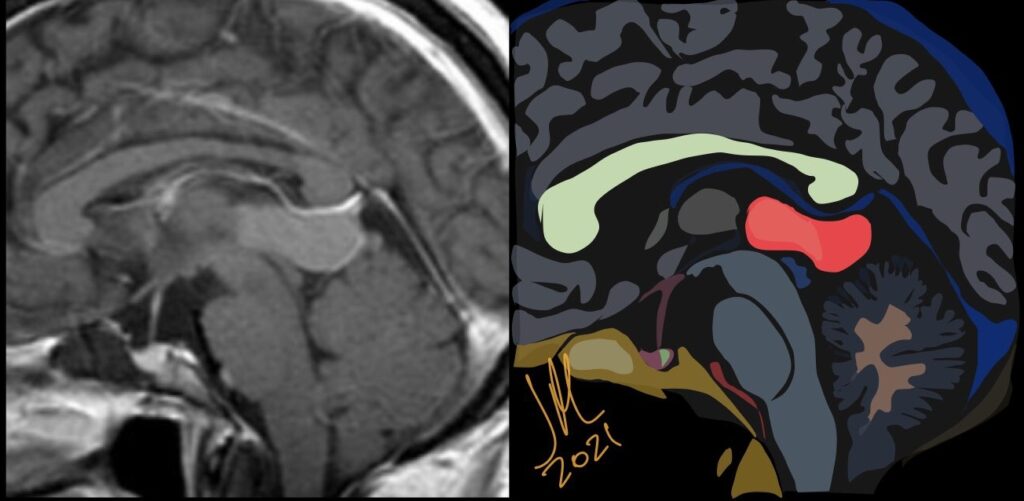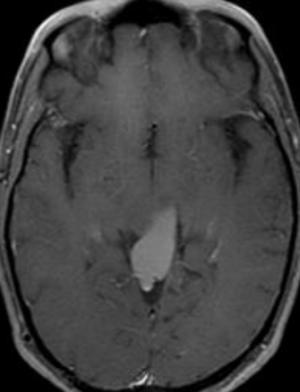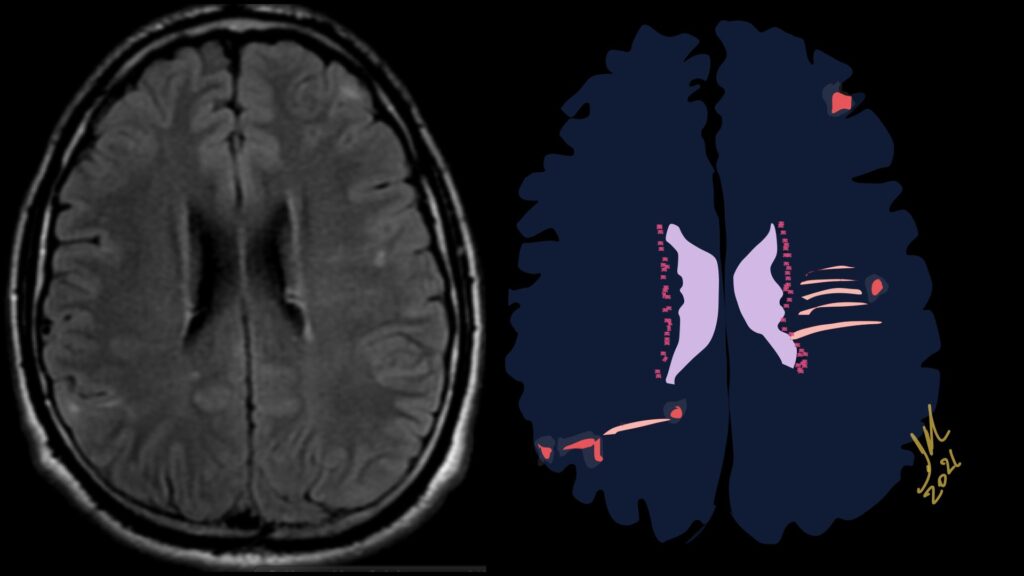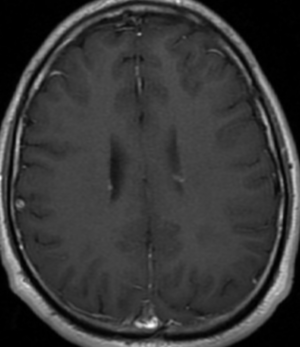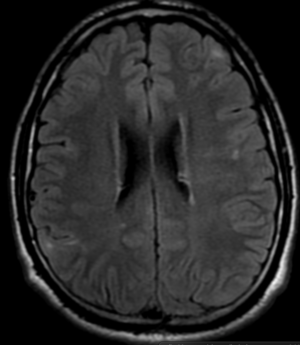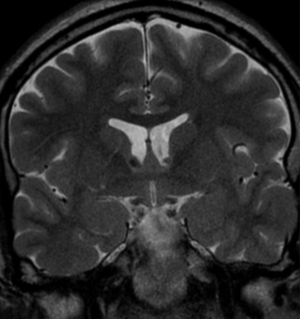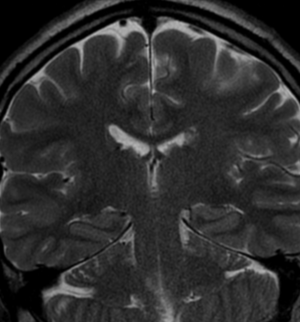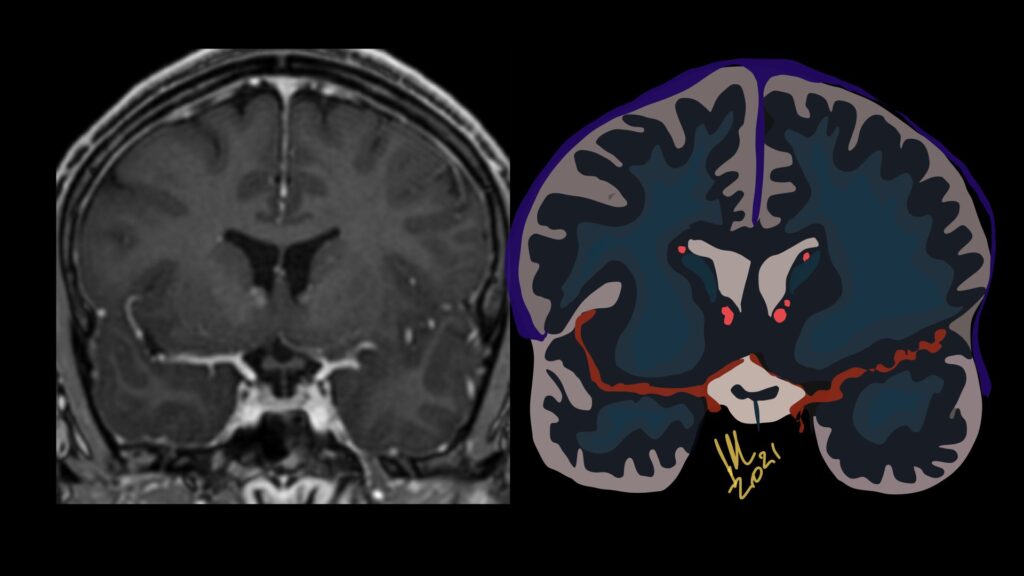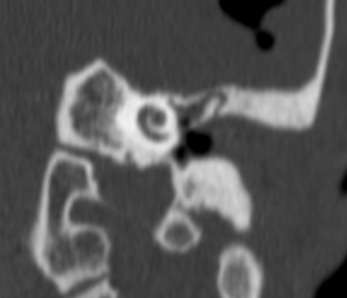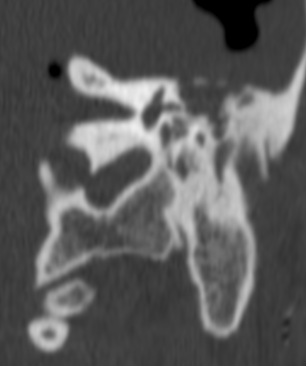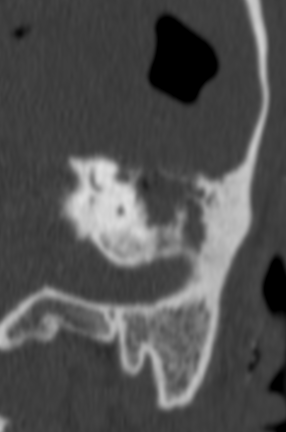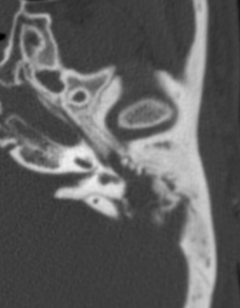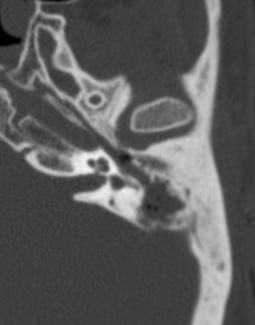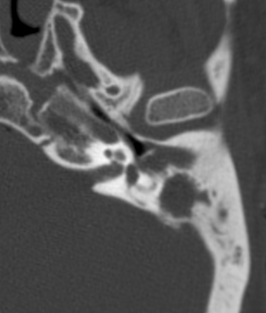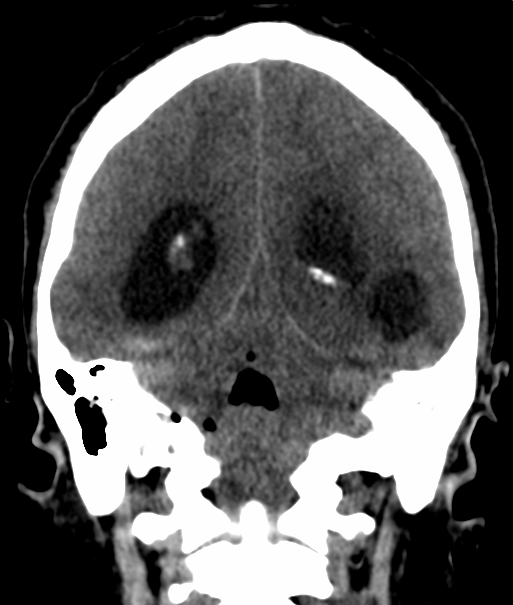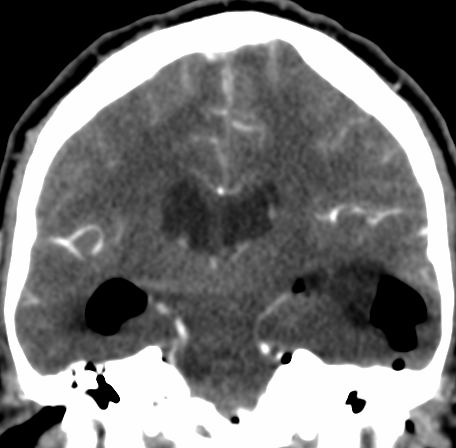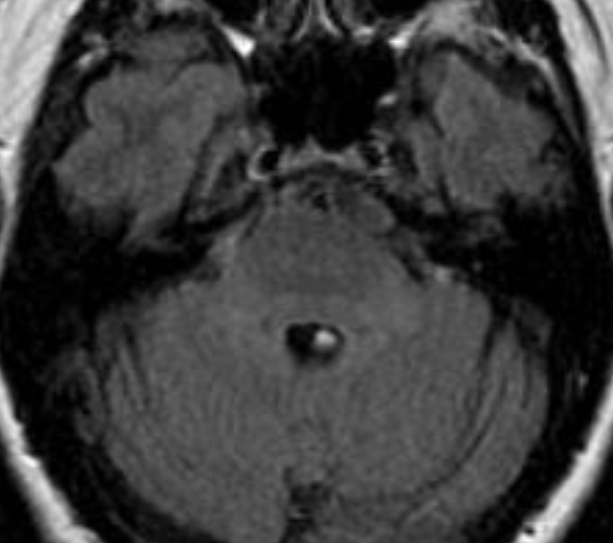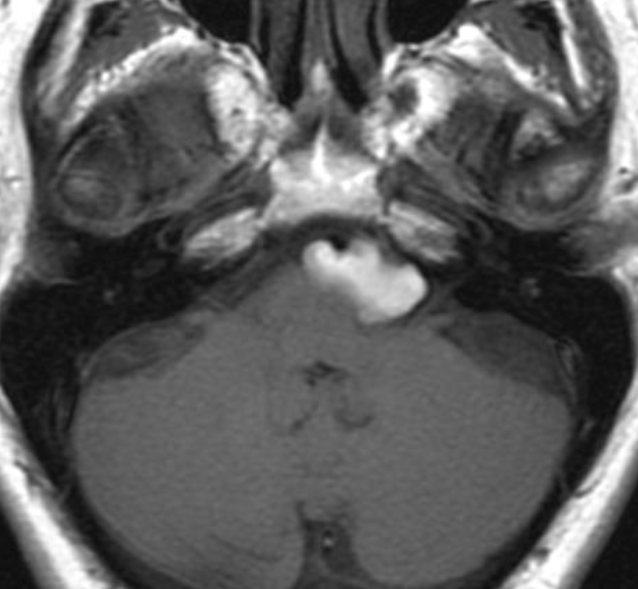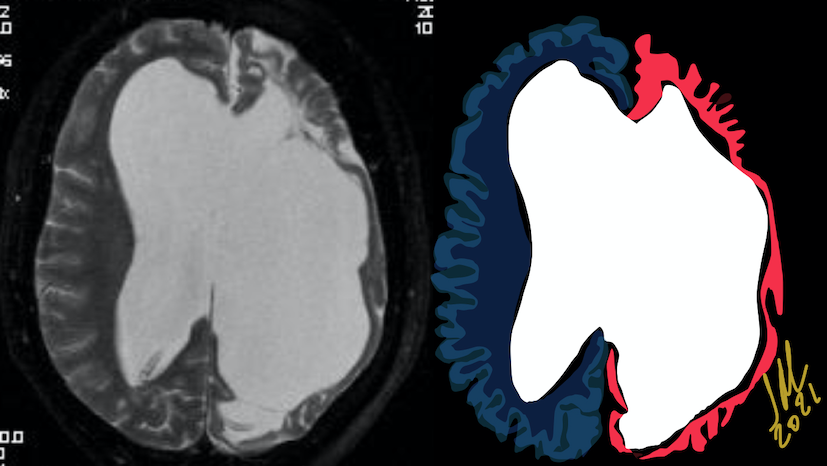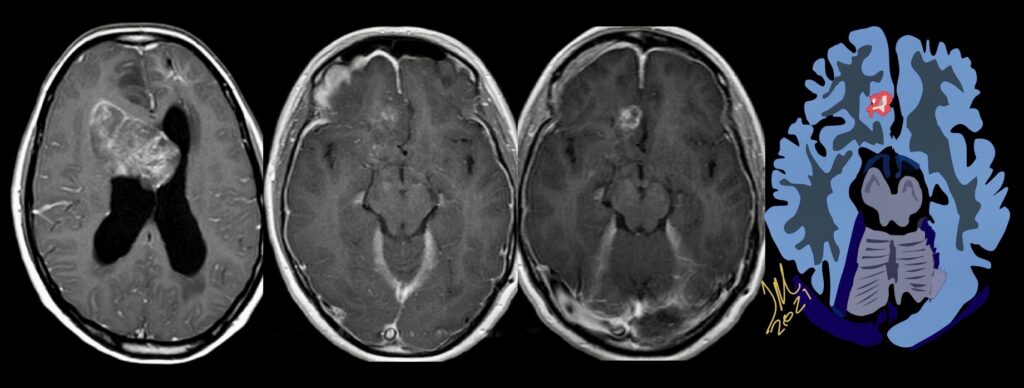
Pseudoprogression is a subacute treatment-related effect with MRI features mimicking those of tumor progression. Patients can present with an increase in contrast enhancement and peritumoral edema at MRI. The diagnosis of pseudoprogression is typically made retrospectively on the basis of spontaneous improvement or stabilization of imaging findings without intervention.
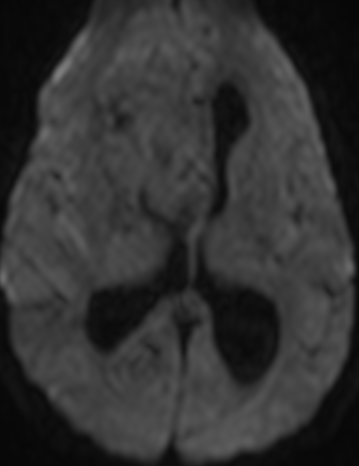
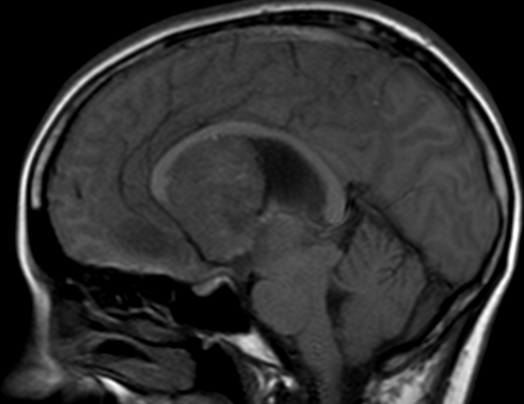
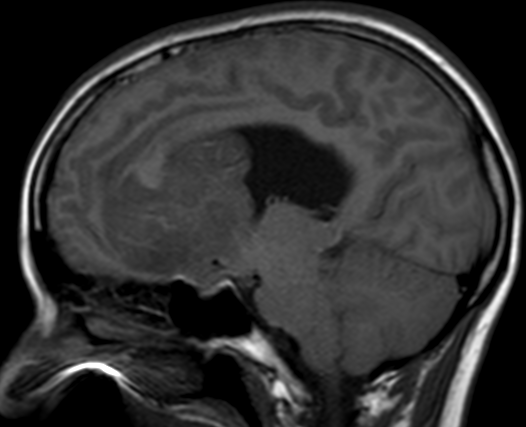
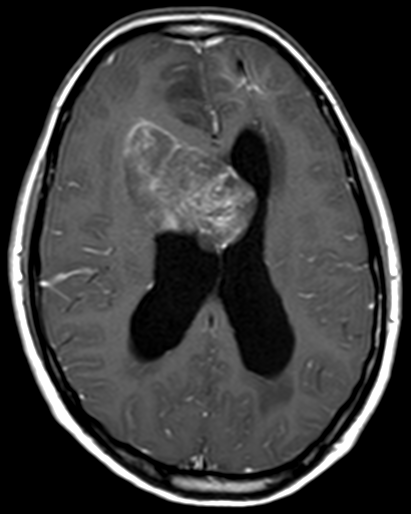
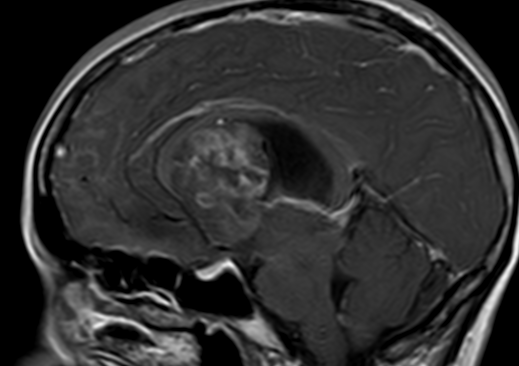
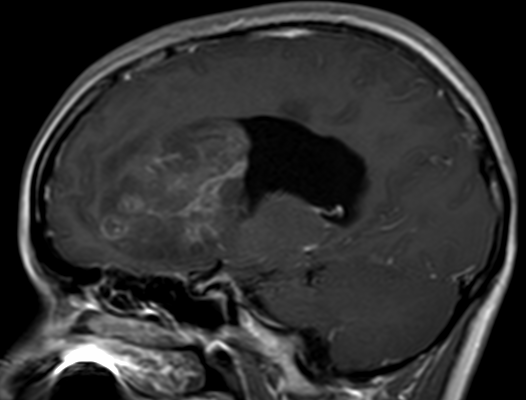
Pseudoprogression typically develops in the setting of combined RT and temozolomide therapy for high-grade or low-grade glioma. but is also observed with immune checkpoint inhibitors in combination with RT for brain metastases and in cases where chemotherapy-infused wafers were placed in the surgical cavity. Pseudoprogression usually develops within 3 months after completion of chemoradiation therapy and is often clinically asymptomatic. The timing of pseudoprogression is earlier than the typical period in which radiation necrosis is described after RT alone; therefore, it is often classified as an early delayed reaction to radiation. Pseudoprogression is most likely induced by a marked local tissue reaction with an inflammatory component, edema, and abnormal vessel permeability causing new or increased enhancement at MRI. Pathologically, pseudoprogression is found to correspond to gliosis and reactive radiation-induced changes without evidence of viable tumor
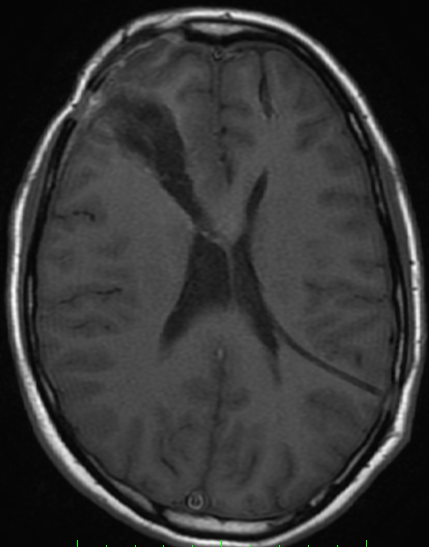
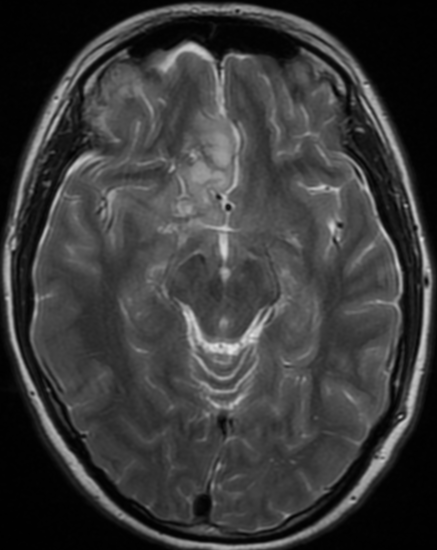
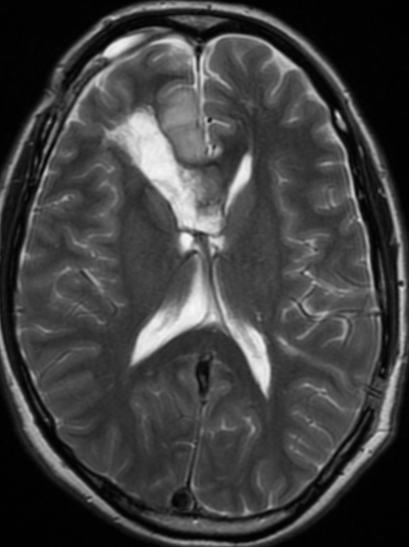
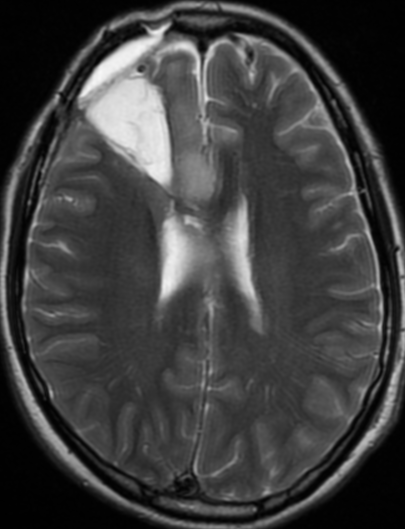
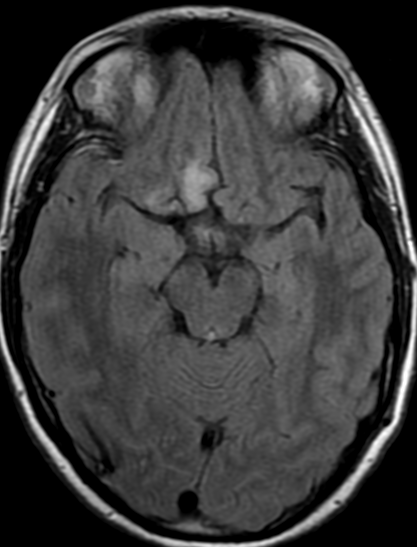
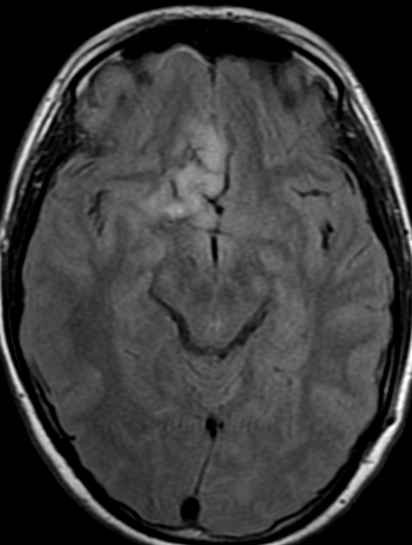
The only method of distinguishing pseudoprogression and true tumor progression is to perform follow-up examinations of the patient because conventional MRI does not allow differentiation of the two conditions. Imaging may be regularly performed at 2–3-month intervals throughout the follow-up period although the frequency of imaging can be variable across institutions.
In clinical practice, the following features can be helpful: (a) presence of symptoms and (b) methylation status of the MGMT gene promoter. REF;Nov 20 2020 https://doi.org/10.1148/rg.2021200064
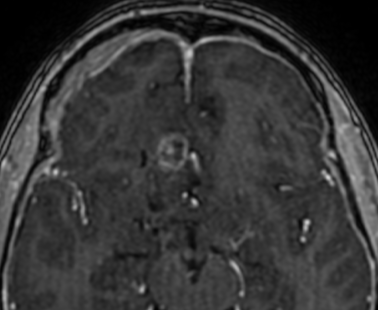
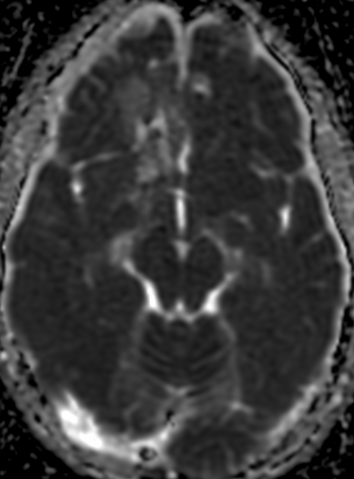
The current standard treatment protocol for glioblastomas is surgical resection followed by 6 weeks of radiation therapy plus concomitant temozolomide chemotherapy (CCRT) and 6 cycles of adjuvant temozolomide chemotherapy. This protocol increases median survival from 12 to 15 months. Tumors with hypermethylation of the O6-methylguanine-DNA methyltransferase promoter gene show pseudoprogression more frequently. Enlarged enhancing lesions on conventional MR images may actually represent pseudoprogression in up to 46.8%–64% of cases.
The Response Assessment in Neuro-Oncology (RANO) Working Group proposed that within the first 12 weeks of completion of radiation therapy, when pseudoprogression is most prevalent, tumor progression can only be determined if most of the new enhancement is outside the radiation field or if there is pathologic confirmation of progressive disease.
MR imaging techniques such as DWI and dynamic susceptibility contrast PWI. On DWI, ADC values are higher in necrotic tissue than in recurrent tumor tissue because of the high cellularity of tumor tissue. However, the use of DWI is limited due to the heterogeneity of tumor content. Reduced diffusion represents not only highly cellular tumor areas but also inflammatory processes.
On PWI, high relative cerebral blood volume (rCBV) is considered active neovascularization and viable tumor. rCBV > 1.47 had 81.5% sensitivity and 77.8% specificity for differentiating pseudoprogression from tumor progression. However, rCBV analysis has limitations because most lesions have variable tumor fractions; therefore, mean rCBV and histogram-based metrics may be influenced by the rCBV from both tumoral and nontumoral components. Reference : https://doi.org/10.3174/ajnr.A3876
the appearance of enhancing lesions on MR imaging within the first 6 months after completion of chemoradiation therapy poses a challenge because it can reflect true progression (TP) or treatment-related changes known as pseudoprogression (PsP). PsP occurs in approximately a third of all patients with glioblastoma. Accurate identification of PsP and TP is critical because patients with TP may require a change in therapeutic strategy while those with PsP may not. The heterogeneity and variability in response did not allow differentiating TP from PsP simply by visual inspection of the parametric maps. However, a quantitative analysis of DTI parameters and rCBVmax from the enhancing regions of the lesion demonstrated better assessment of treatment response in patients with glioblastomas.
Identification of TP
Early identification of TP could prevent further delays in repeat surgery or enrollment in alternative clinical trials. The LRM analysis indicated that the best model to distinguish TP from PsP or mixed responses was based on FA, CL, and rCBVmax. Higher anisotropy values have been reported in glioblastomas compared with brain metastases and primary cerebral lymphomas. High FA in glioblastomas is probably related to the orientation of overproduced extracellular matrix.
Identification of PsP
Accurate identification of PsP is critical for patient management because unnecessary repeat surgery/biopsy can be avoided in these patients and they can continue on an effective temozolomide regimen with standard imaging follow-up of 3–6 months, thereby reducing patient care costs. Logistic regression analysis showed that the best model to differentiate PsP from TP and mixed response included FA and rCBVmax.
Pseudoprogression is predominantly a subacute treatment-related reaction. Pathologically, it corresponds to gliosis and radiation-induced reactive changes including disruption of the BBB, inflammation, increased permeability, and edema. These changes cause increased enhancement on MR imaging and can mimic TP. combination of FA and rCBVmax can help in identifying PsP from TP or mixed response.
Identification of Mixed Response
On a practical level, posttreatment new enhancing lesions usually contain a mixture of viable neoplasm and treatment-induced changes, and a more accurate assessment of the relative contribution of each entity can guide clinical decision-making. However, most previous studies have attempted to only differentiate between PsP and TP. REF; American Journal of Neuroradiology January 2016, 37 (1) 28-36; DOI: https://doi.org/10.3174/ajnr.A4474