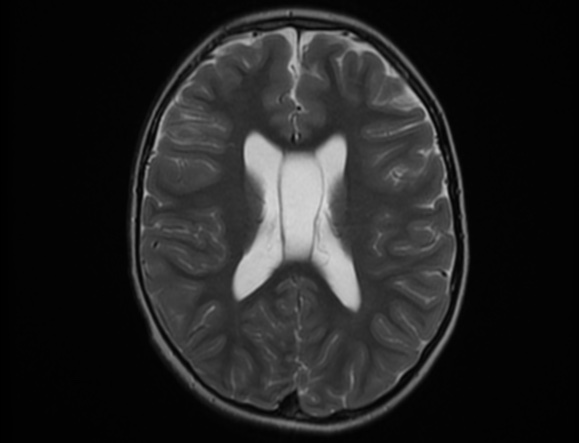
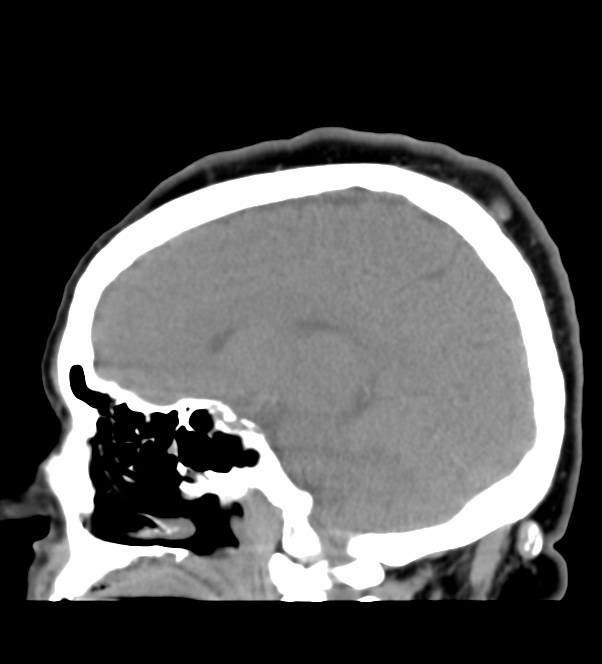
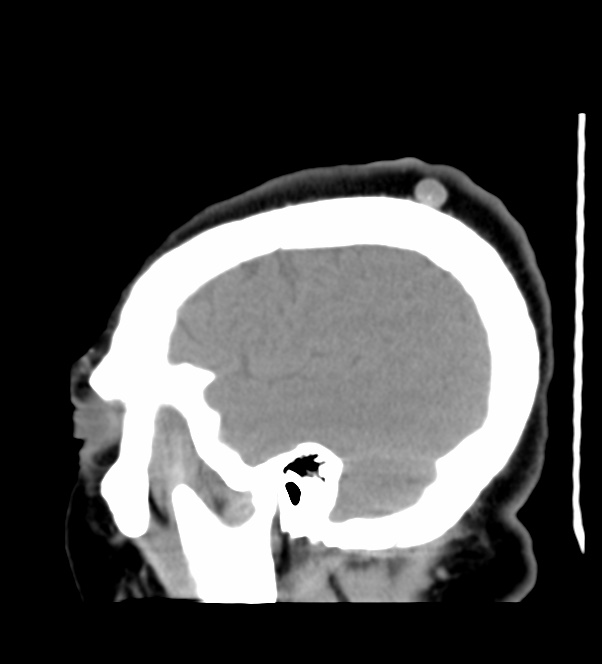
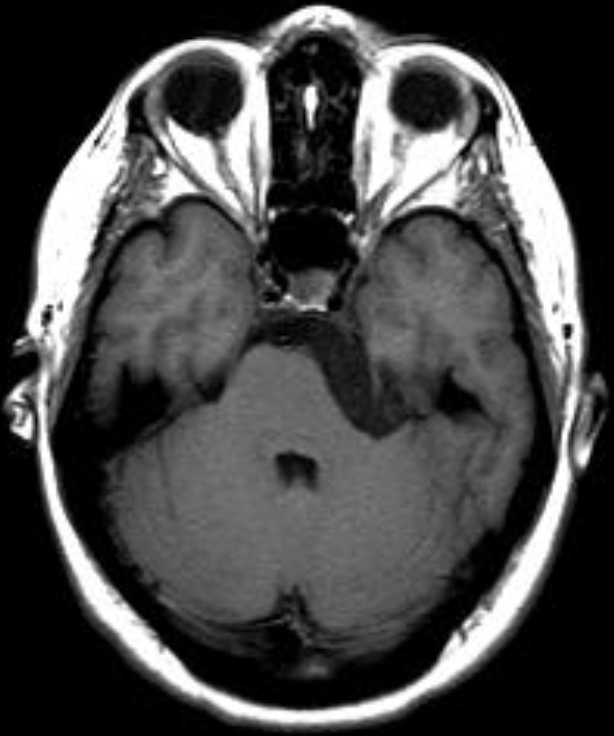
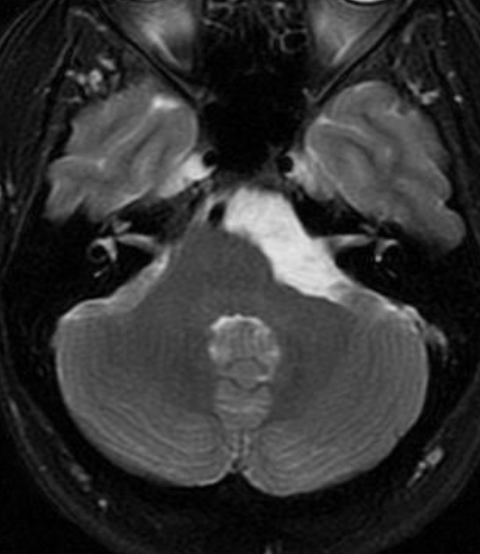
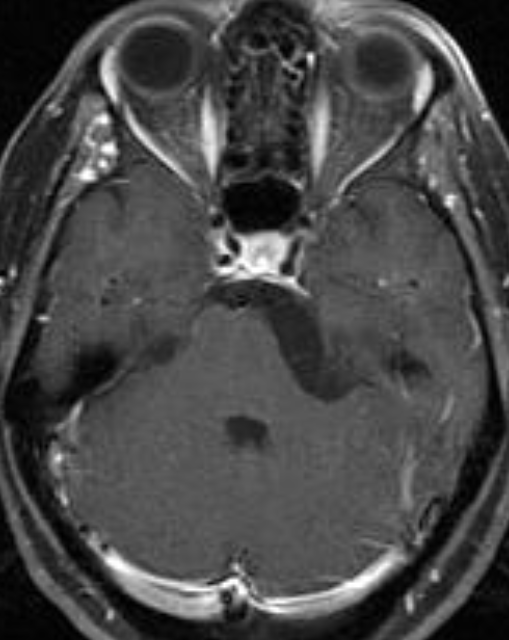
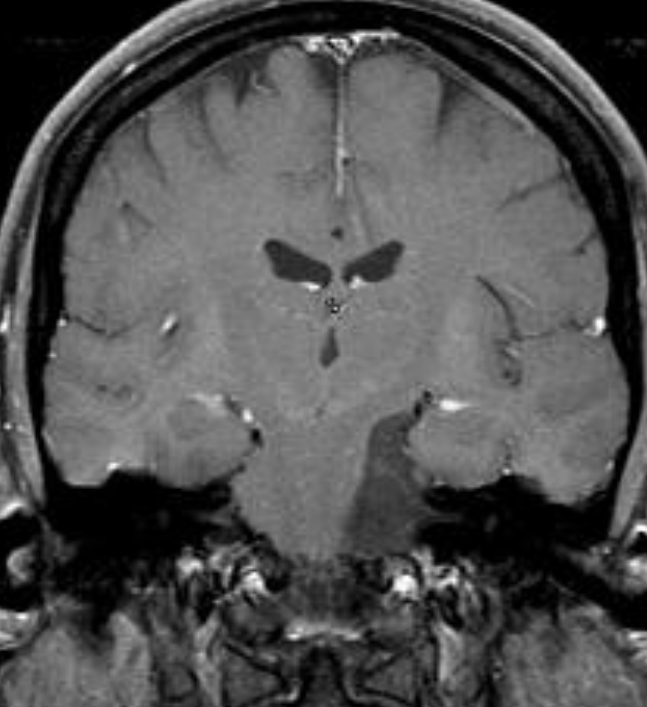
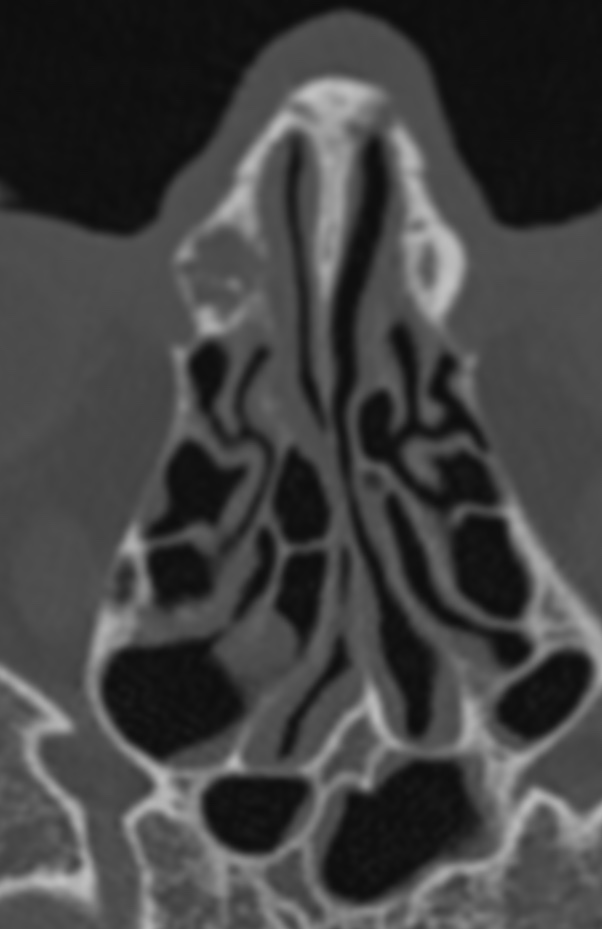
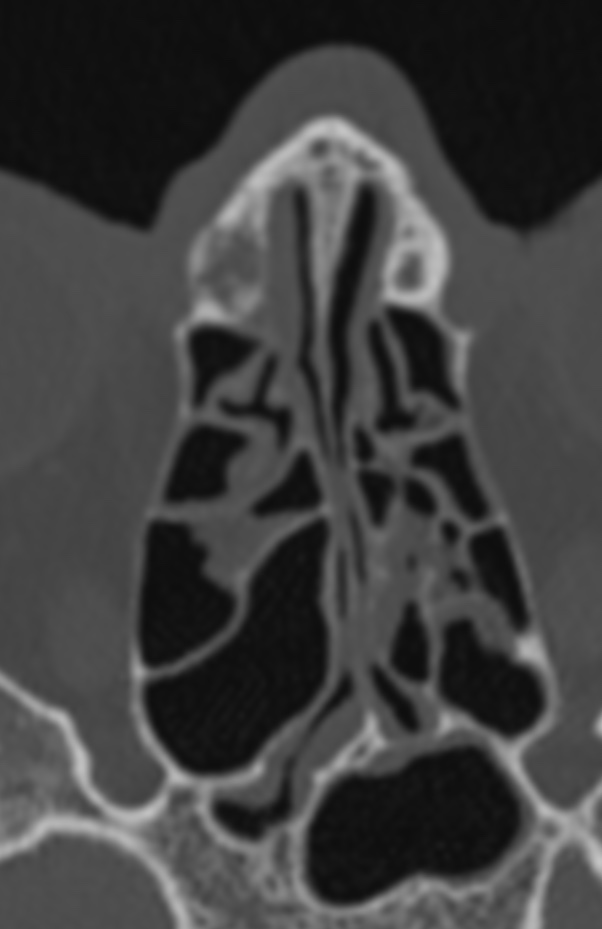
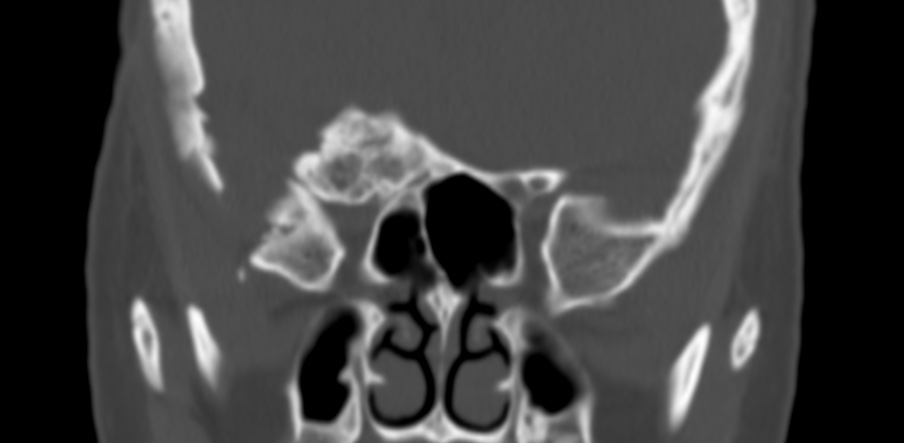
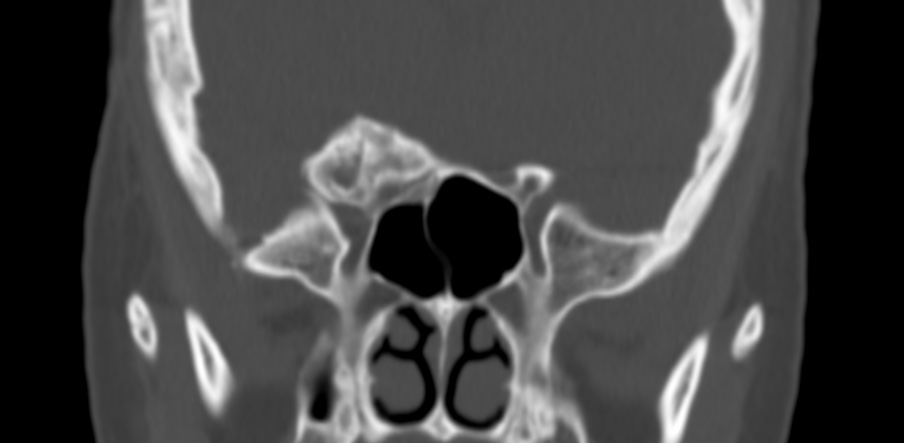
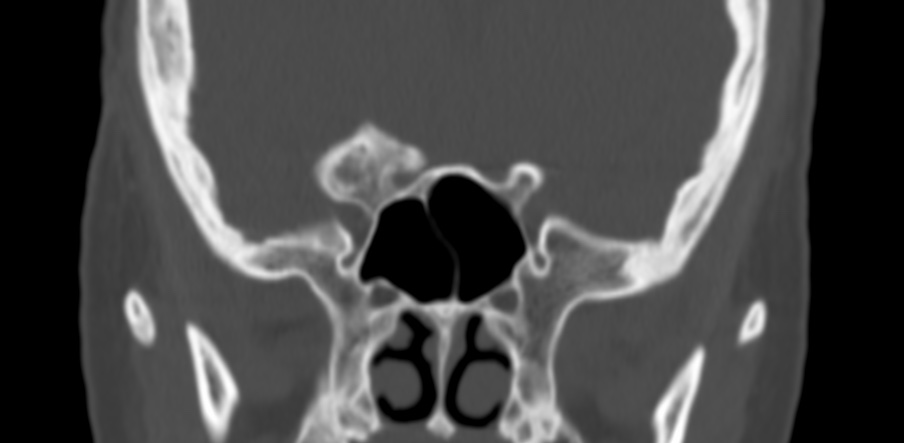
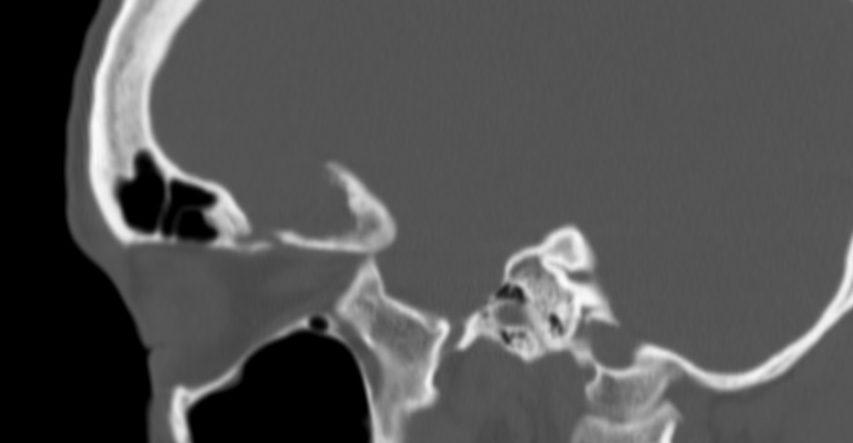
The septum pellucidum is a structure that is marginated by the corpus callosum and body of the fornix. It is composed of white matter leaves along the medial walls of the lateral ventricles and is lined by ependyma along its ventricular surfaces . The entire space between the leaves of the septi pellucidi is the cavum septi pellucidi et vergae, with the space anterior to either the foramina of Monro or an arbitrary vertical plane formed by the forniceal columns being the CSP and the space posterior being the cavum vergae.
When the two leaves fail to fuse and form the septum pellucidum, a number of postnatal anatomic variants can result including the CSP, cavum vergae, and cavum veli interpositi. The CSP and the cavum vergae usually freely communicate with one another. Usually a cavum vergae is seen in association with a CSP, although it can also occasionally be seen in isolation . In general, persistence of the CSP in postnatal life is considered a normal variant.
The leaves of the septi pellucidi begin to close at approximately 6 months’ gestational age from back to front. Nearly all term infants have closure of the cavum vergae and the majority of infants 3–6 months old have closure of the cavum septi as well . The cavum sometimes persists into adulthood as a normal variant, typically small, measuring less than 4 mm in transverse diameter, in healthy individuals . The terminology can be confusing. In general, when the two leaves are separated, this may be referred to as the “cavum septi pellucidi” or “CSP”; when the leaves are fused to form a single structure, it is referred to as the “septum pellucidum”
The usual clinical presentation of PTT is that of a long-standing, subcutaneous, cystic nodule that slowly progresses to a large, nodular mass, often following a history of trauma or inflammation. The tumor preferentially arises in areas of dense hair follicle concentrations, and about 90% of cases occur on the scalp, with the residual 10% occurring mainly on the back. Other, less common locations include the vulva, nose, mons pubis, buttock, wrist, chest, and elbow.
Women are affected in more than 80% of cases, and the average age of patients is 65 years . The presentation is nearly always that of a single lesion, but, rarely, multiple lesions are seen.
Areas of necrosis, calcification, and hyalinization may be seen. The stroma is usually fibrous and shows a variable inflammatory reaction, including foreign body giant cells.
Although PTT is generally considered biologically benign, even though histologically indistinguishable from squamous cell carcinoma in some cases, malignant PTT has been reported. Saida et al suggested three stages in the oncologic development of a malignant PTT: the adenomatous stage of the trichilemmal cyst, the epitheliomatous stage of the PTT, and the carcinomatous stage of the malignant PTT. A rare occurrence of PTT with spindle cell carcinoma has also been reported .
Reference:
American Journal of Neuroradiology January 2001, 22 (1) 180-183;
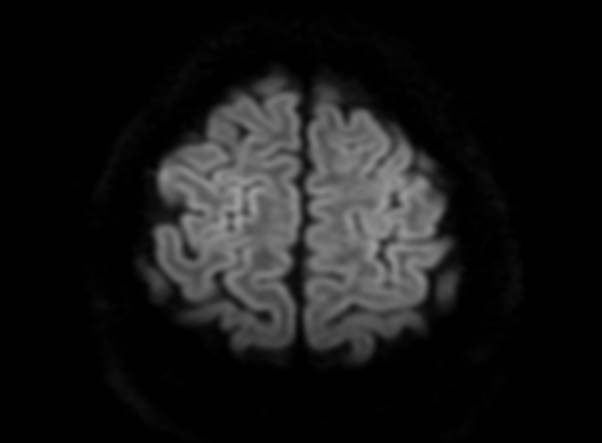
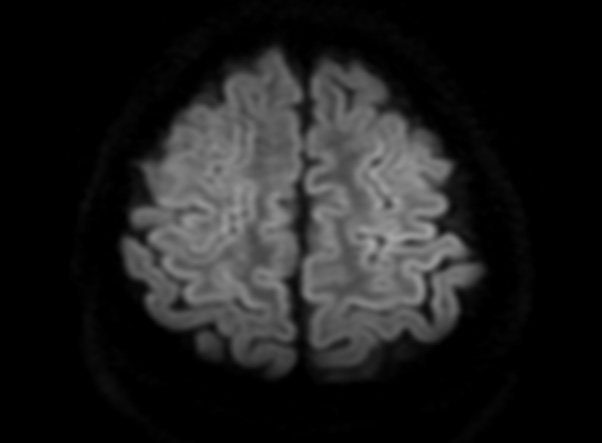
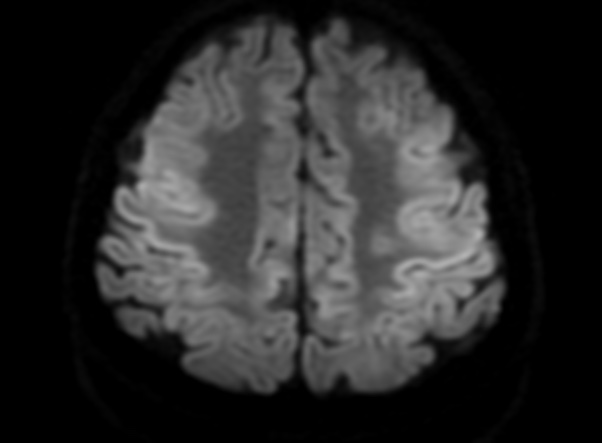
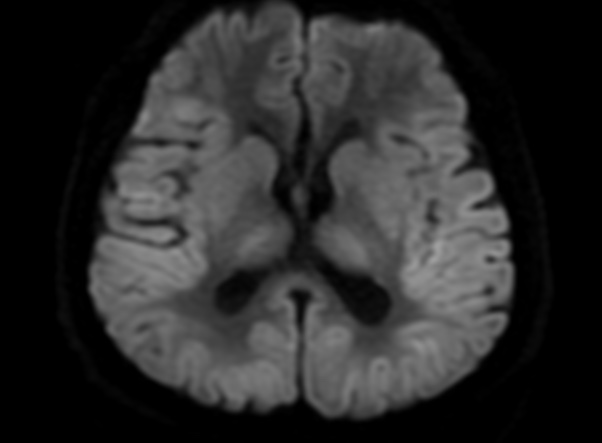
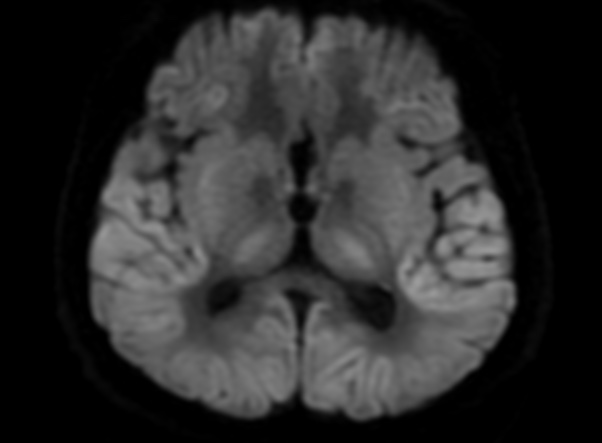
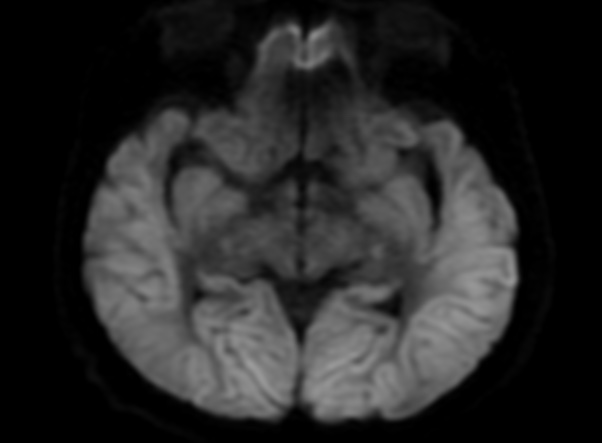
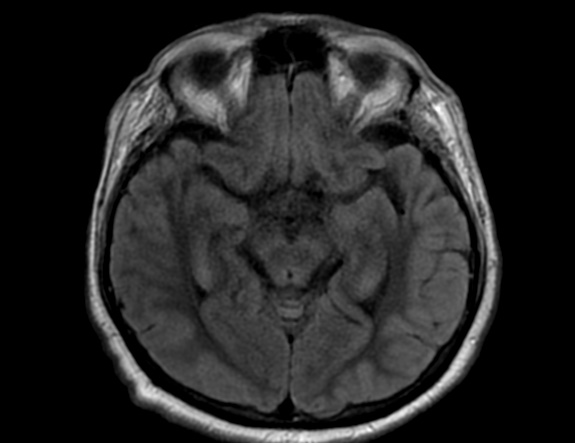
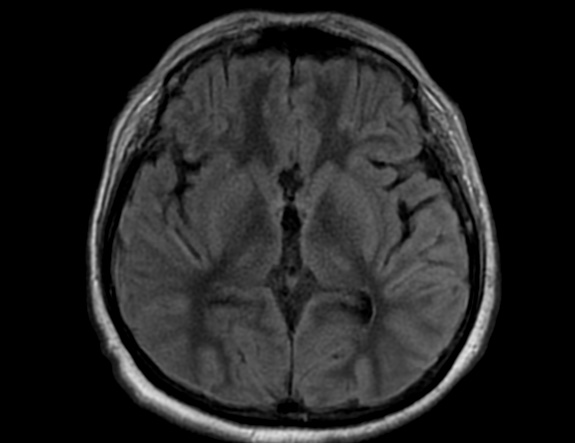
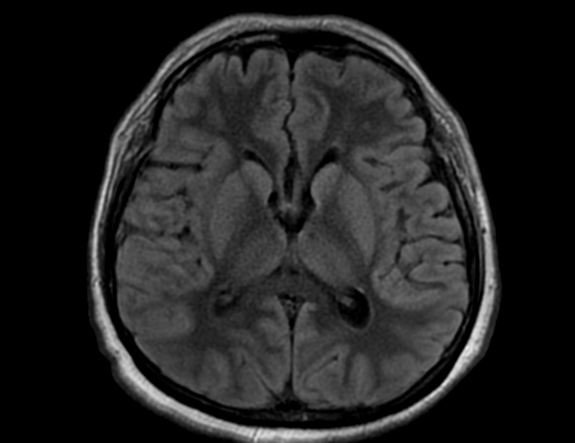
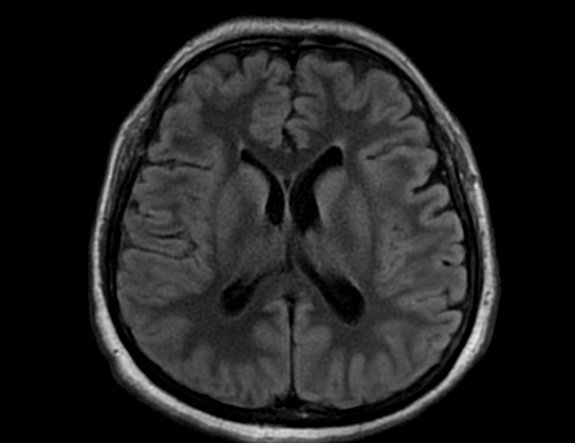
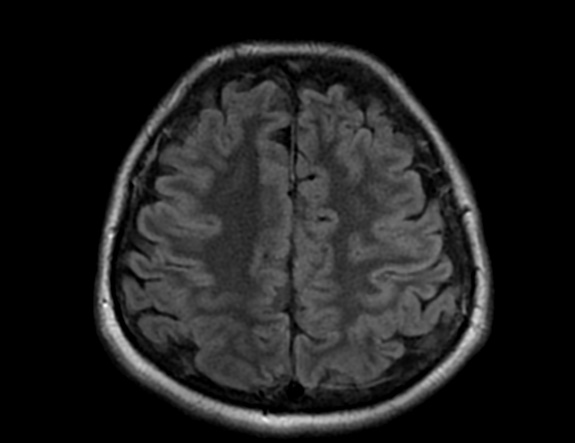
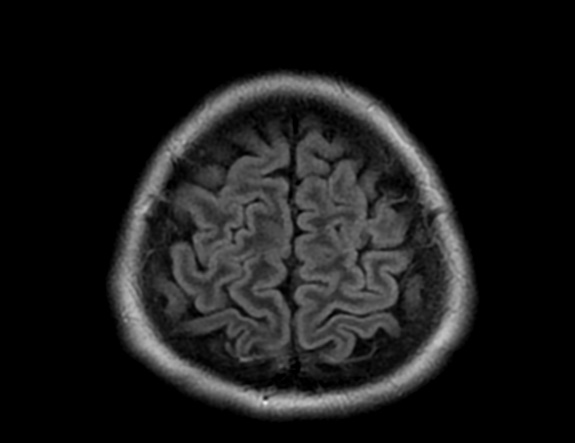
FLAIR image show symmetric hyperintense lesions in the parietal cortices bilaterally.
Diabetic ketoacidosis with prolonged hyperglycemia may cause subtle FLAIR and diffusion abnormalities in the cortex associated with elevations in glucose, myoinositol, taurine, and ketones in MR spectroscopy.
Diabetes mellitus type 1 (DM1) is one of the most common chronic pediatric illnesses. Diabetic ketoacidosis (DKA) is serious complication in children with DM1 and has an increasing incidence. DKA is a state of severe insulin deficiency, either absolute or relative, resulting in hyperglycemia, ketonemia, acidemia, and systemic inflammation.
DKA is a rather common presentation or complication of pediatric DM1 patients and occurs in about 36% of diabetic children younger than 5 years of age and about 16% of teenagers older than 14 years. In the majority of pediatric patients, DKA has an excellent prognosis. In about 1% of the children DKA results in acute morphologic and functional brain changes that are associated with poor long-term neurocognitive outcome. Neuroimaging plays a key role in the early diagnosis of diabetic children with DKA and acute CNS involvement. Accordingly, it important that neuroradiologists are aware of the possible neuroimaging findings associated with DKA.
Cerebral edema is the most common neuroimaging finding associated with pediatric DKA and occurs in about 0.5%–1% of pediatric DKA.
Cerebral edema in DKA is associated with increased morbidity and mortality in up to 50% of affected children and occurs almost exclusively in pediatric patients. Children with more severe acidosis, hypocapnia, and dehydration have a higher risk of developing cerebral edema. The exact pathomechanism of cerebral edema in DKA, however, remains unclear and is most likely multifactorial. Possible pathomechanisms include (1) osmotic gradients drawing fluid into hypertonic astrocytes during intravenous therapy, (2) intracellular accumulation of sodium and water as a correction of intracellular acidosis due to the accumulation of lactate, free fatty acids, and ketone bodies, and (3) vasodilatation and reperfusion following cerebral vasoconstriction due to hypoocapnia.
On CT and MRI, cerebral edema in children with DKA is characterized by effacement of the sulci and basilar cisternal spaces (especially the suprasellar, quadrigeminal plate and ambient cisterns), compression and decreased size of the cerebral ventricles, and reduction of the gray-white matter differentiation.
Diffusion-weighted and diffusion-tensor imaging may further characterize cerebral edema and differentiate between vasogenic and cytotoxic edema. Increased ADC values representing vasogenic edema have been shown in the basal ganglia, thalamus, frontal white matter, and periaqueductal gray matter of children with DKA and cerebral edema. Increase in ADC values correlates with the degree of dehydration and hyperventilation at presentation, but not with factors related to initial osmolality or osmotic changes during treatment.These findings suggest that cerebral hypoperfusion may be a key role in the pathomechanism of cerebral edema in DKA. The role of cerebral hypoperfusion is supported by perfusion-weighted imaging studies showing shorter mean transit times and higher peak tracer concentrations, again suggesting vasogenic edema.
Severe cerebral edema may cause complications with high morbidity and mortality such as focal stroke and subfalcine or transtentorial herniation. Focal infarction secondary to cerebral edema may occur in up to 20% of children with DKA and typically involves the mesial basal ganglia, thalamus, periaqueductal gray matter, and dorsal pontine nuclei. Brain herniation is an uncommon complication of cerebral edema in DKA that typically occurs 6–13 hours after onset of symptoms and is clinically characterized by sudden deterioration of consciousness, absent brainstem responses, and eventual respiratory arrest. Compression of the major vessels of the circle of Willis due to downward herniation may play an additional significant factor in the occurrence of focal ischemic strokes.
Other possible neuroimaging findings in DKA include ischemic and hemorrhagic stroke, sinovenous thrombosis, and extrapontine myelinolysis. Ischemic stroke in DKA may occur independently from cerebral edema and be due to increased blood viscosity and thrombosis due to hyperosmolality, systemic inflammatory reaction, vascular endothelial injury, acidemia-induced red blood cell rigidity, and hyperglycemia-induced vasoconstriction. Hemorrhagic stroke is less common in acute DKA and may be secondary to sinovenous thrombosis.
In addition, hyperglycemia and acidosis could enhance endothelial damage with subsequent extravasation of red blood cells through the leaky vessels and increase the risk of hemorrhagic transformation of ischemic strokes.
Finally, extrapontine myelinolysis is a very rare complication. Extrapontine myelinolysis is characterized by T2-/FLAIR-hyperintense signal and restricted diffusion in the claustrum and putamen with inconsistent involvement of the hippocampi and subcortical white matter and may be associated with pontine myelinolysis. The exact pathomechanism of extrapontine myelinolysis in DKA is unknown, but is most likely related to electrolyte imbalance in the context of DKA and its rapid correction.
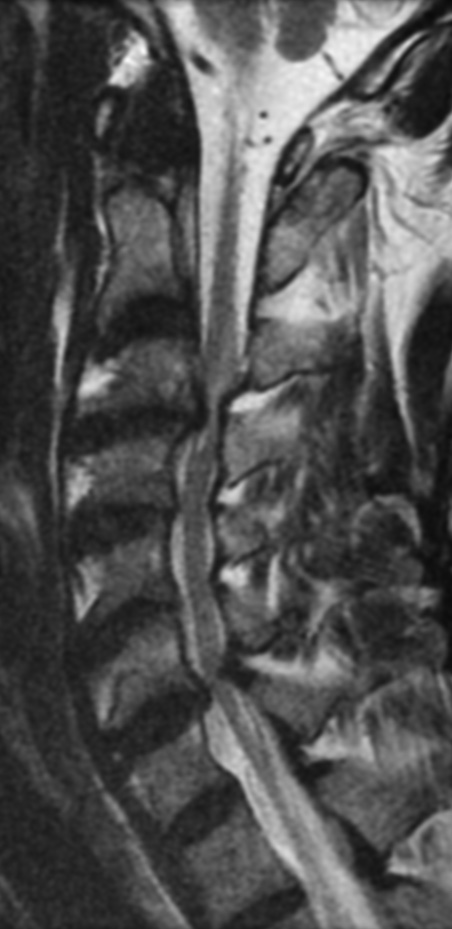
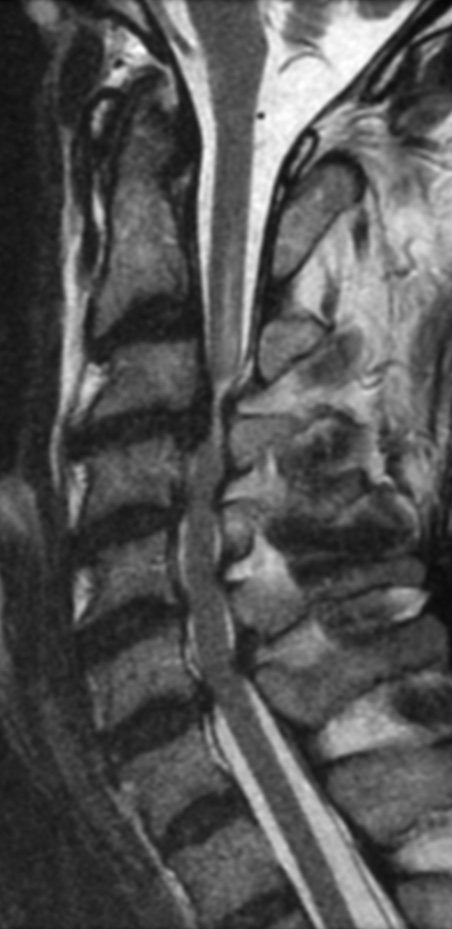
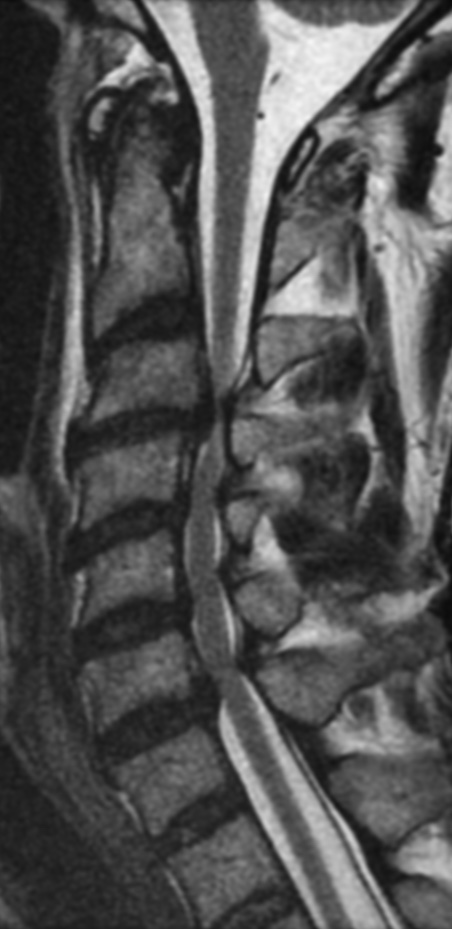
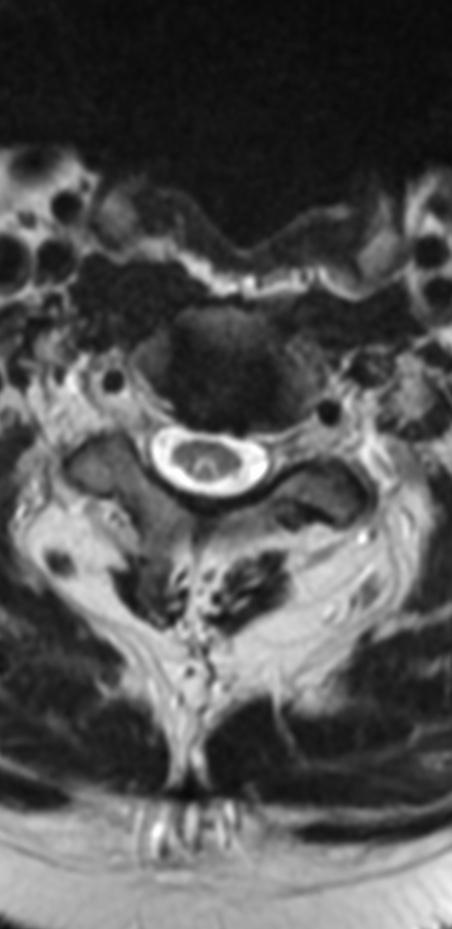
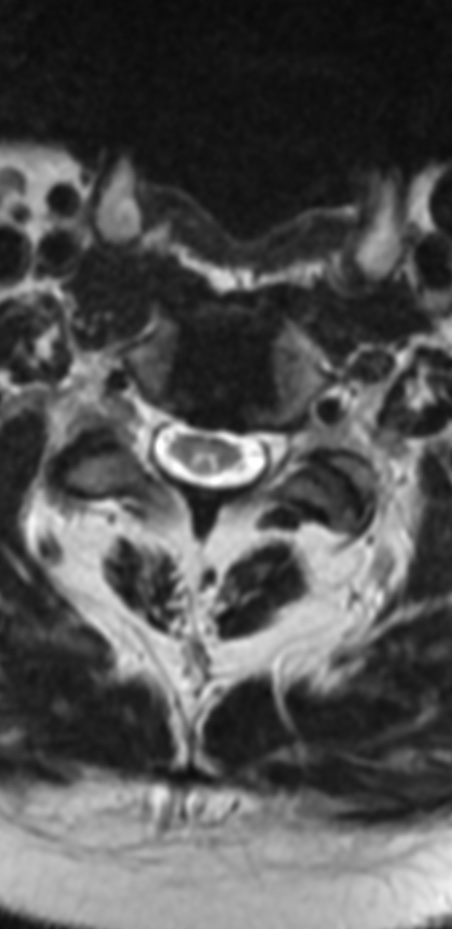
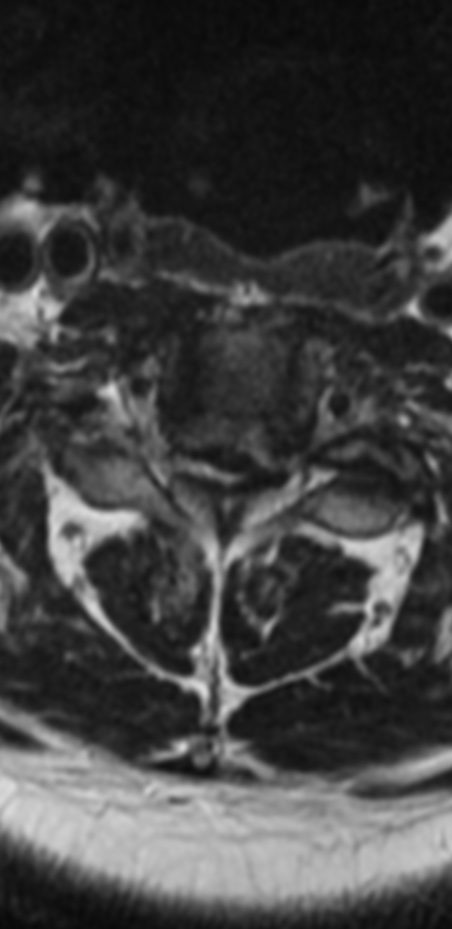
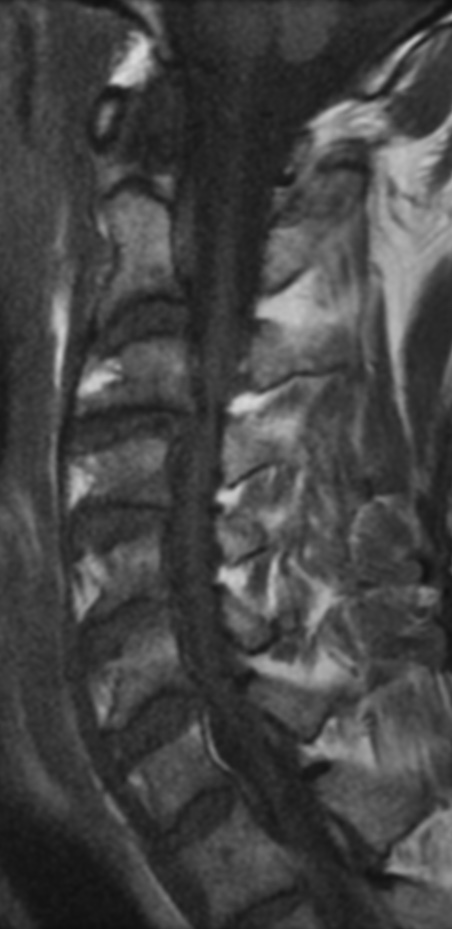
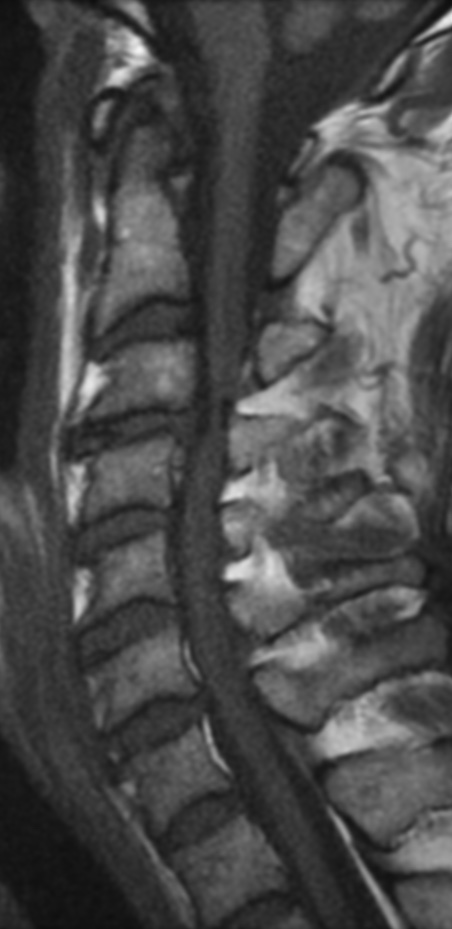
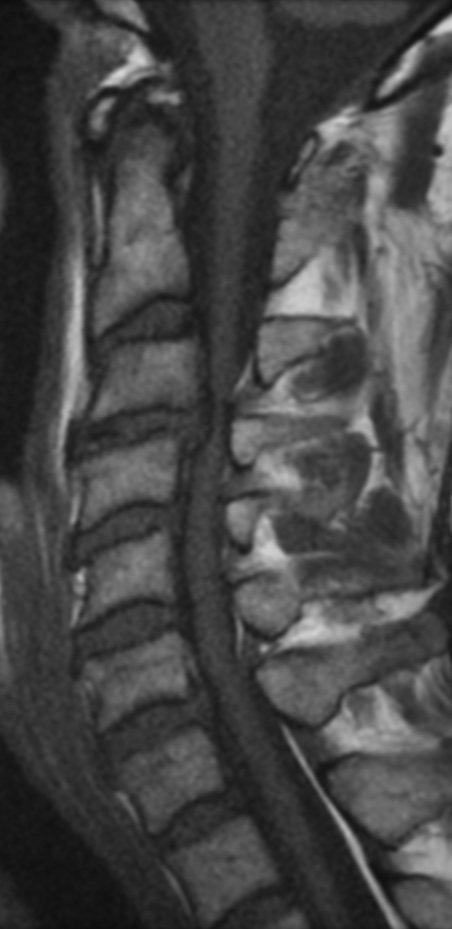
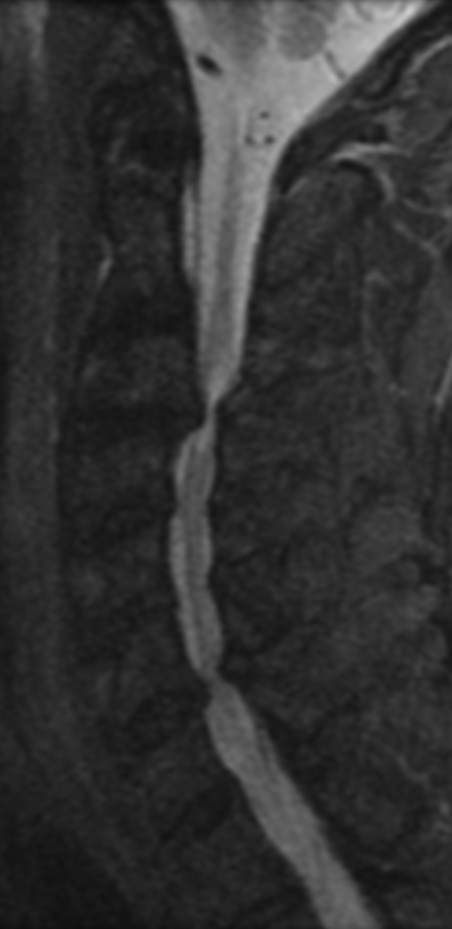
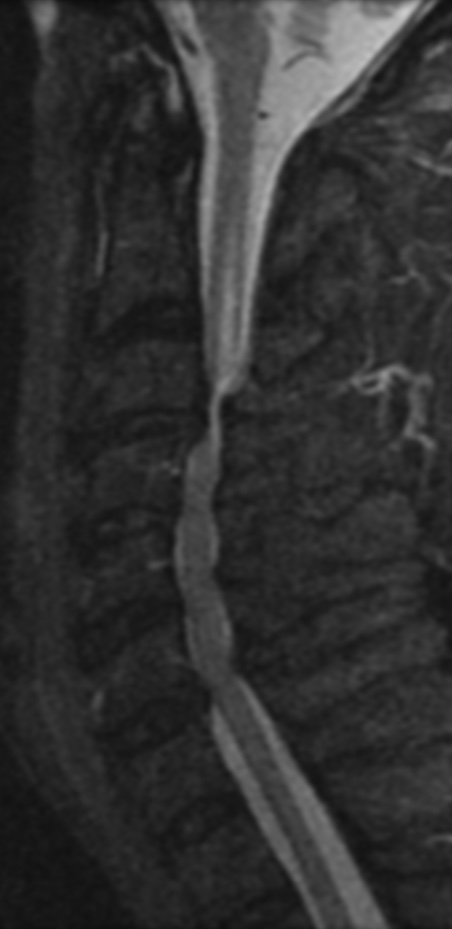
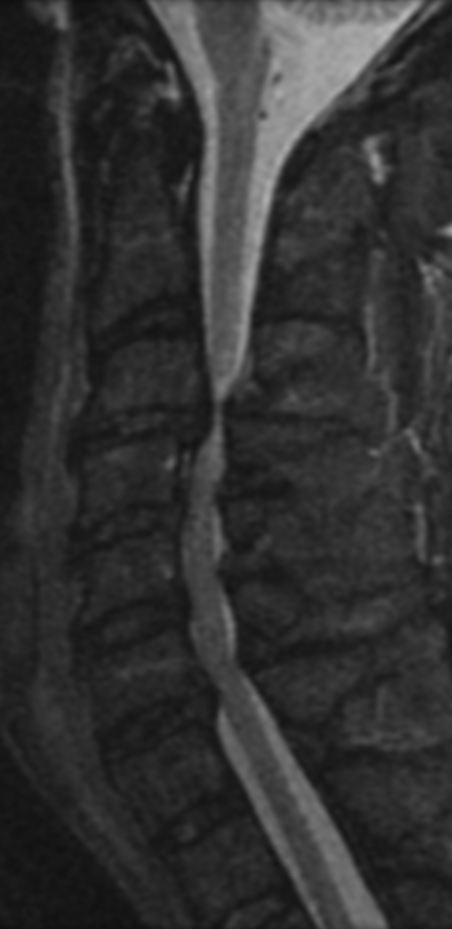
Cervical myelopathy is a condition describing a compression of the spinal cord at the cervical level of the spinal column resulting in spasticity , hyperreflexia, pathologic reflexes, digit/hand clumsiness, and/or gait disturbance. Classically it has an insidious onset progressing in a stepwise manner with functional decline.
An MRI can provide some guidance for clinicians and patients about the potential for improvement. Based on a systematic review of MRI findings by Tetreault et al. in 2013:
High-intensity changes on T2 and low intensity on T1: poorer recovery rate, worse motor symptom improvements
More frequent high signal intensity on T2 predicts worse recovery.
MRI: Canal space of less than 10 mm indicates stenosis. Best to evaluate cord and disk space. Of note, up to 19% of asymptomatic patients have major cervical abnormalities which can be misleading. An MRI is imperative to evaluate spine pathologies. Compression and signal changes on T1 and T2 are indicative of myelopathy pathologies.
Cervical spondylotic myelopathy will frequently involve compression of the lateral corticospinal tracts resulting in voluntary skeletal muscle control, and the spinocerebellar tracts proprioception. Together, these deficits are responsible for the wide-based spastic gait with clumsy upper extremity function that is classic to cervical myelopathy. Additional commonly involved spinal cord regions are the spinothalamic tracts, which are responsible for contralateral pain and temperature sensation, the posterior columns, which are responsible for the ipsilateral position and vibration sense, and the dorsal nerve root, which is responsible for dermatomal sensation
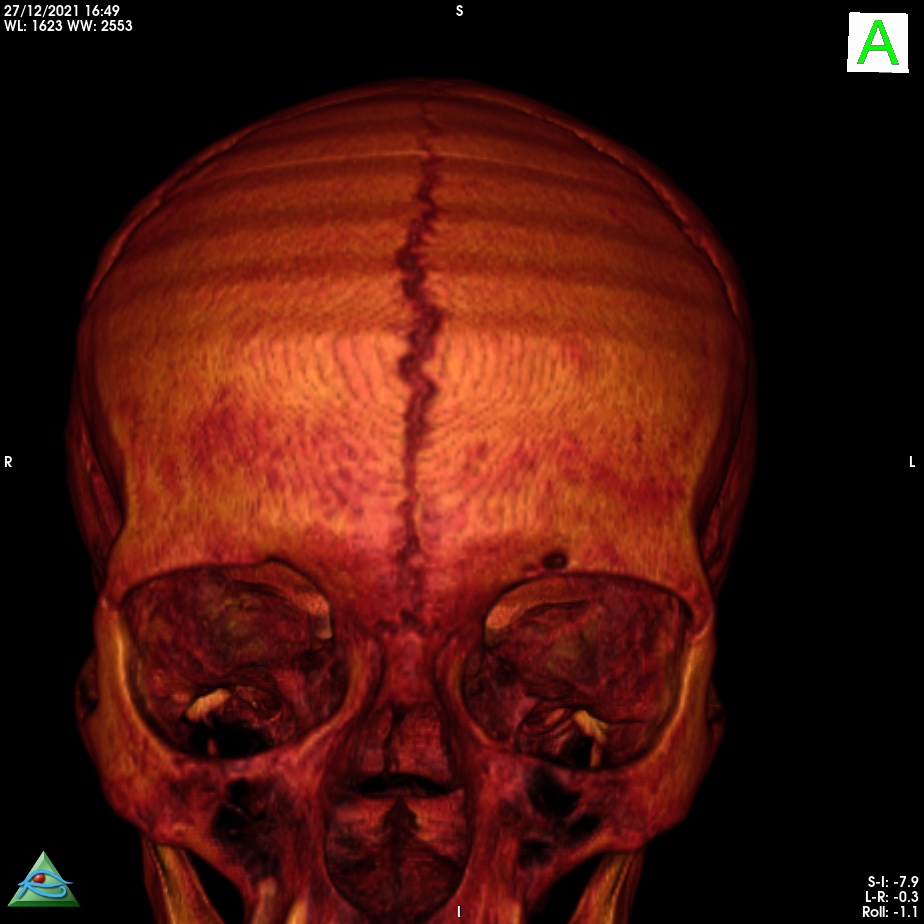
The persistence of the metopic suture is called metopism. This suture disappears by the second or third year of life. It is thought to be a normal variant of the cranial sutures . It forms from the lack of union of the two frontal bones during embryonic development.
Metopism is found in approximately 5% of Asians and 9% of European Caucasians and 1% of Blacks.
Epidermoid tumors arise from ectoderm trapped within/displaced into the central nervous system. They show predilection for CPA Angle (up to 40%), 4th ventricle, suprasellar region, and spinal cord. They are the 3rd most common CPA tumor, comprising approximately 7% of CPA pathology. CPA lesions can produce 5th and 7–12th cranial nerve neuropathies.
Recurrent episodes of aseptic meningitis caused by cyst content rupture may occur. Symptoms include fever, meningeal irritation, and hydrocephalus. A 26-year-old female presented with headaches. Head magnetic resonance imaging (MRI) revealed right CPA tumor with brain stem compression. There was evidence of restricted diffusion in diffusion-weighted imaging, typical of epidermoid tumor.
The tumor grows slowly and is soft and very pliable, conforming to the shape of the adjacent brain and CSF spaces in which it is growing. The cysts, frequently referred to as “pearly tumors” because of their gross external appearance, account for approximately 0.2% to 1.8% of brain tumors. Although congenital in nature, they usually do not present clinically until the third or fourth decade. The cysts may be extradural or intradural in location. The intradural lesions are frequently located in the cisterns of the cerebellopontine angle, supra- and parasellar regions, and middle cranial fossa, as well as the cisterna magna. Tumors may also develop in the tela choroidea, usually in the temporal horn of the lateral ventricle but occasionally in the fourth ventricle.
On CT the lesions are hypodense and do not enhance with contrast material. They are difficult to differentiate from arachnoid cysts based on their density; however, their external surface is usually lobulated in configuration compared with the smooth surface of an arachnoid cyst. There may occasionally be focal calcifications in their walls. In contrast to arachnoid cysts, epidermoids tend to engulf and surround vessels and cranial nerves, whereas arachnoid cysts displace such structures.
On T1-weighted MR images, epidermoid tumors demonstrate subtle hypointensity compared to CSF. There is usually mild inhomogeneity of low intensity, with some patchy regions of isointensity within the lesion. On T2-weighted sequences the tumors show marked hyperintensity similar to or greater than that of CSF, with significant heterogeneity of the signal.
The low-intensity signals within the tumor hyperintense pattern are probably the result of the cellular debris and solid cholesterol crystals within the cysts . A high-intensity rim may surround the portion of the cyst on T2-weighted sequences that probably represents a CSF cleft. The tumors may be dumbbell shaped in configuration and extend from the middle cranial fossa into the posterior fossa. They may have a smooth external surface but in most instances demonstrate a lobulated appearance. No free fatty acids have been found in epidermoid cysts that are of low intensity on the T1-weighted sequences. On MRI, epidermoids are distinctly different from arachnoid cysts, in that epidermoid tumors are virtually never isointense to CSF on MR images when using FLAIR. In addition, diffusion- weighted imaging demonstrates reduced diffusion within epidermoids, simplifying the diagnosis and aiding visualization.
Fibrous dysplasia is a fibro-osseous medullary lesion; it affects a single bone (monostotic) in 70% of cases and more than one bone (polyostotic) in 30% of cases. Polyostotic fibrous dysplasia manifests in association with skin hyperpigmentation and endocrine abnormalities in McCune-Albright syndrome. Fibrous dysplasia in the craniofacial bones accounts for 35% of monostotic fibrous dysplasias, even if it involves several adjacent bones. The skull base and facial bones are common sites of involvement, and in this setting, they may encroach on the nasal cavity. The jaws are the most frequently affected bones. Fibrous dysplasia is more common in the 1st and 2nd decades of life and has a slight female predilection.
The clinical manifestations of fibrous dysplasia include painless swelling, facial deformity, exophthalmos, and symptoms related to narrowing of skull base foramina. Fibrous dysplasias that undergo periods of increased activity can cause increased swelling and discomfort and may mimic osteomyelitis on bone scans. The growth of this lesion tends to decrease after puberty. The diagnosis is confirmed by the presence of the mutation of GNAS 1α, which enables the differentiation of fibrous dysplasia from other fibro-osseous lesions.
The imaging characteristics of fibrous dysplasias depend on the stage of development of the disease and the amount of bone matrix, and vary from lucent to sclerotic lesions. At plain radiography and CT, this lesion has the characteristic ground-glass appearance, which is secondary to the amount of woven bone present. Fibrous dysplasia manifests as an expansile lesion that has ill-defined margins and a thinned cortex; it replaces normal bone but rarely involves bone erosion. CT is key to rendering an accurate diagnosis, given that fibrous dysplasias can exhibit aggressive characteristics at MR imaging, mimicking malignant lesions.
At MR imaging, the appearance of fibrous dysplasia is variable, but it typically has low signal intensity on T1-weighted images. The signal intensity is variable on T2-weighted MR images: it can be low, intermediate, or high. At contrast-enhanced imaging, there is internal heterogeneous enhancement, with the fibrous component typically enhancing more than the osseous component. As a benign lesion, fibrous dysplasia has increased diffusivity on apparent diffusion coefficient maps, which can help in differentiating it from a malignant lesion.
Fibrous dysplasia (FD) is a rare, mosaic disorder in which normal bone and marrow are replaced with fibro-osseous tissue. Expansile lesions lead to deformities, fractures, and disability. FD may involve one or multiple bones, and may occur in association with skin macules and hyperfunctioning endocrinopathies, termed McCune-Albright syndrome (MAS)
Vision loss due to optic neuropathy (ON) is a severely disabling complication of FD. ON is a nonspecific diagnosis referring to damage and atrophy of the optic nerve due to any cause.
There is evidence suggests the mechanism of ON in FD is more complex and likely multifactorial. Since growth hormone excess is an established risk factor for ON in FD/MAS, optic nerve elongation, in the setting of acromegalic macrocephaly, is another proposed mechanism.Furthermore, FD involvement of the facial skeleton leading to asymmetry and proptosis may also stretch the optic nerve leading to ON. Because of this heterogeneity, management of ON in FD is challenging and has been the subject of controversy for decades.
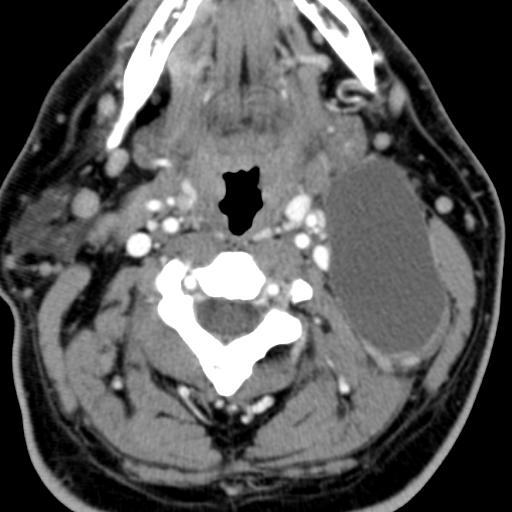
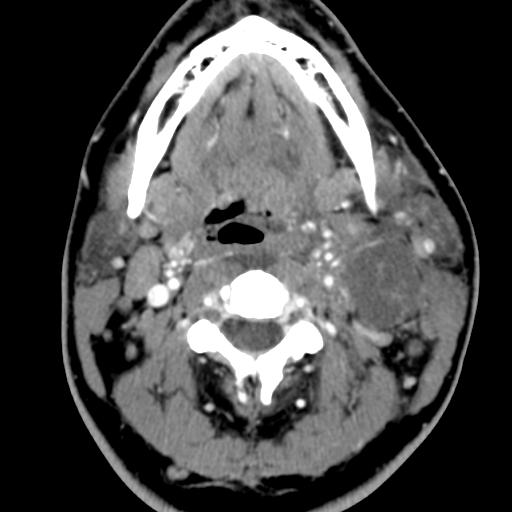
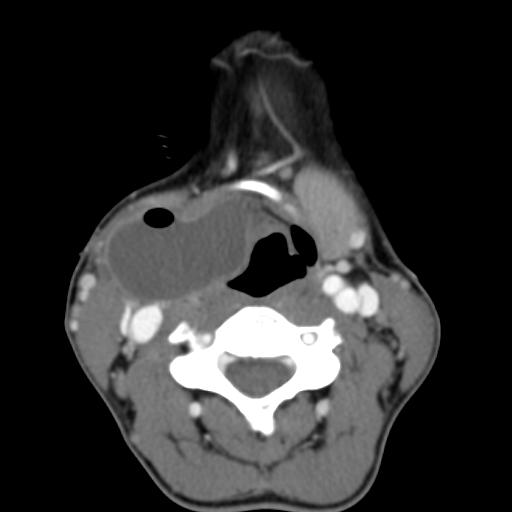
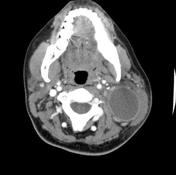
The branchial arches represent the embryological precursors of the face, neck and pharynx. Anomalies of the branchial arches are the second most common congenital lesions of the head and neck in children. They may present as cysts, sinus tracts, fistulae or cartilaginous remnants and present with typical clinical and radiological patterns dependent on which arch is involved.
different cases
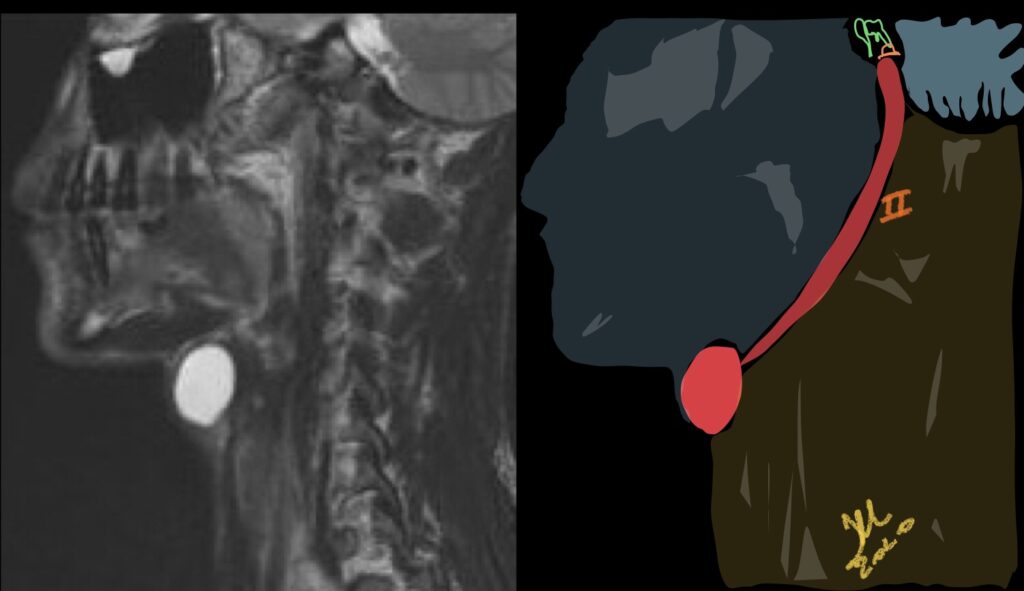
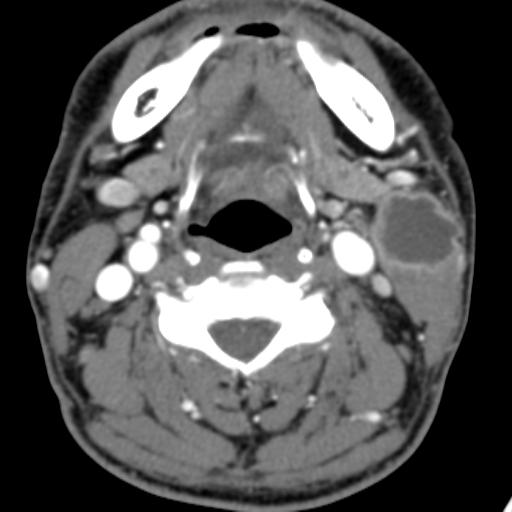
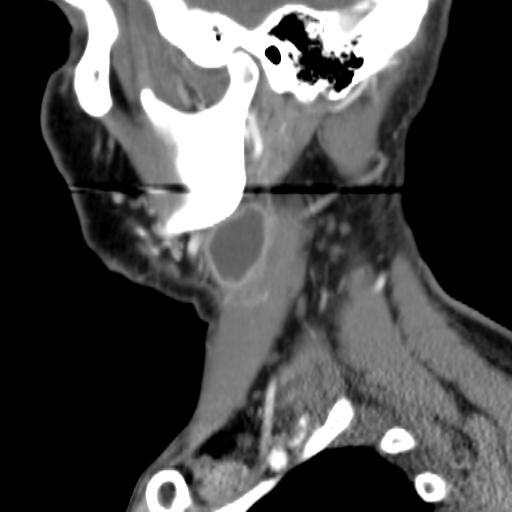
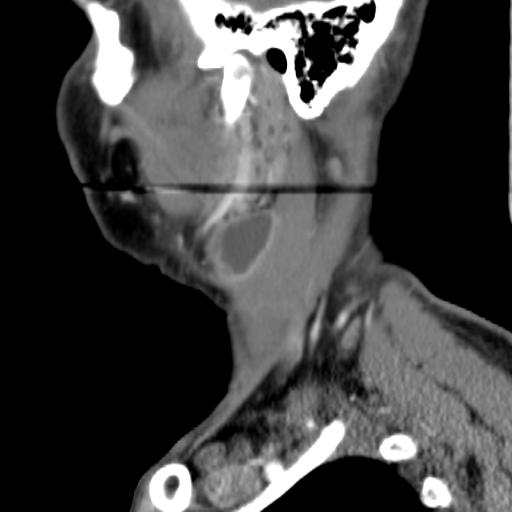
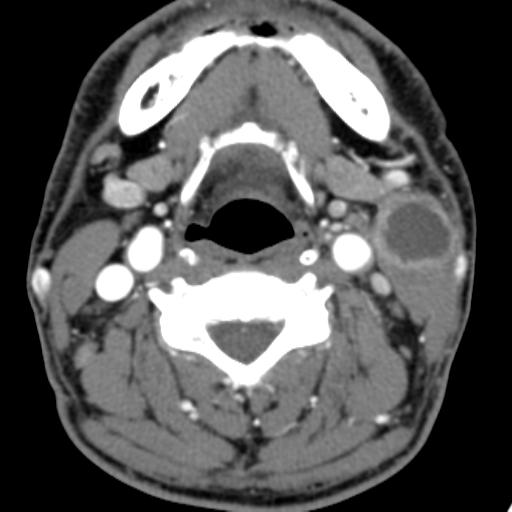
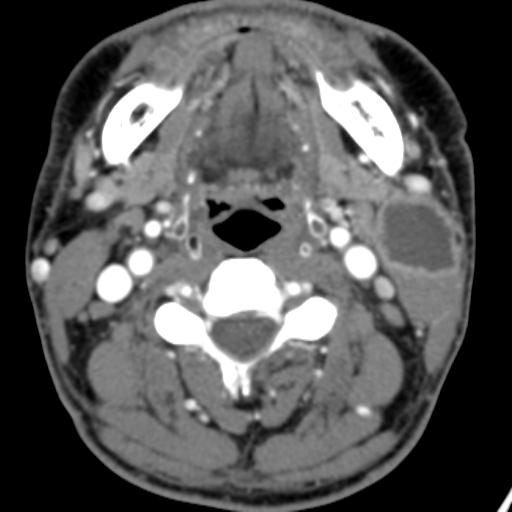
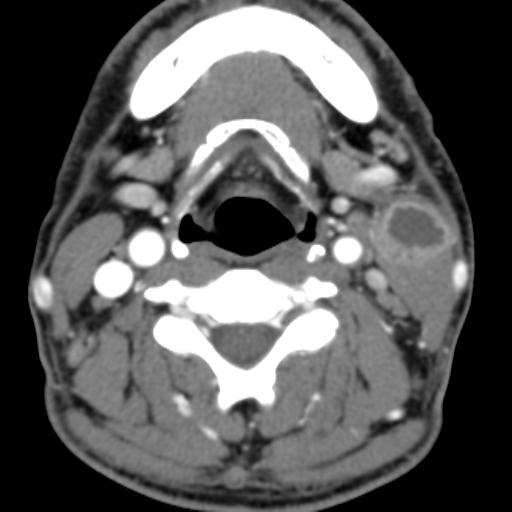
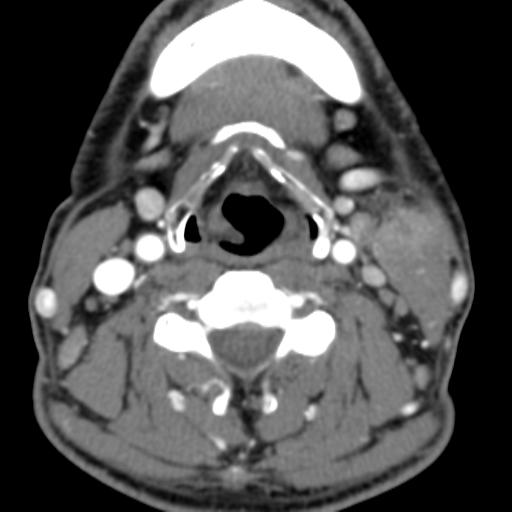
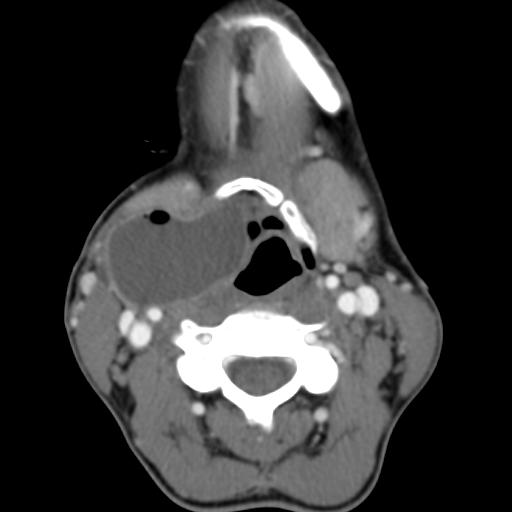
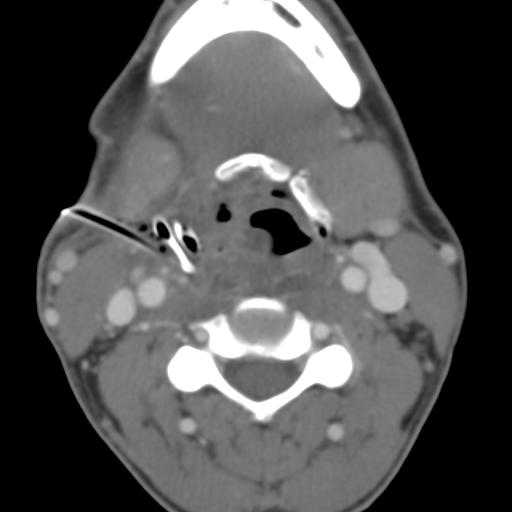
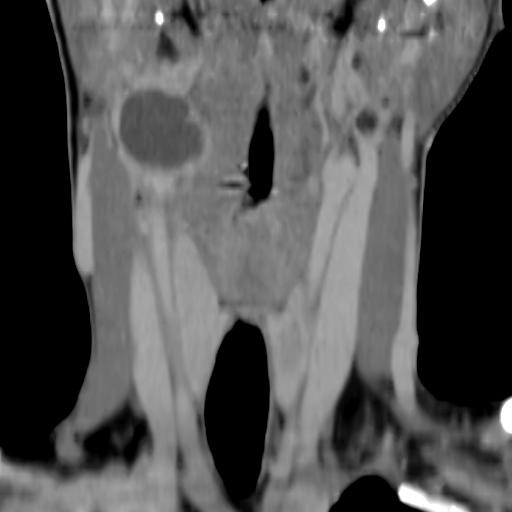
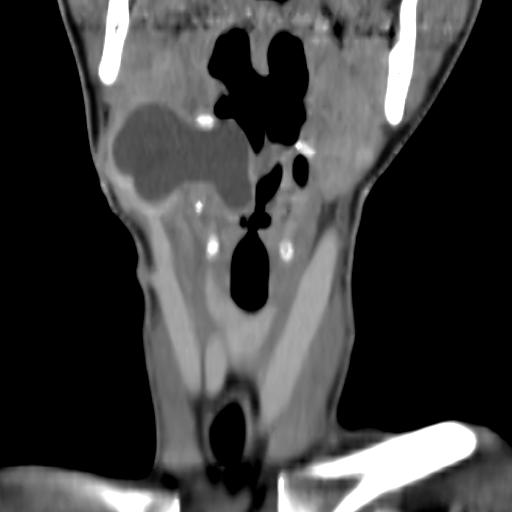
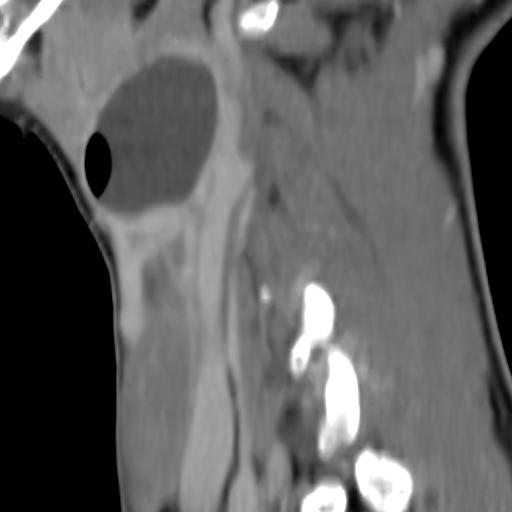
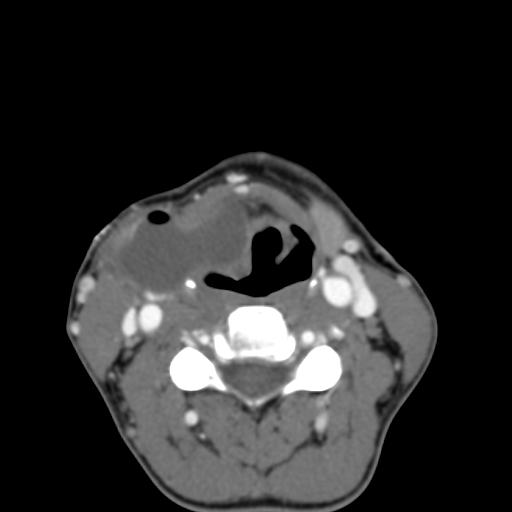
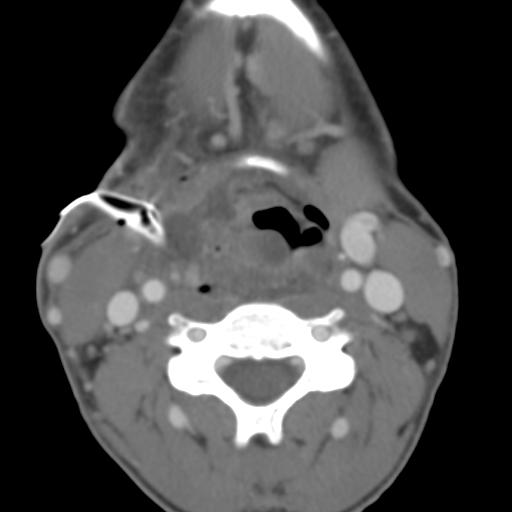
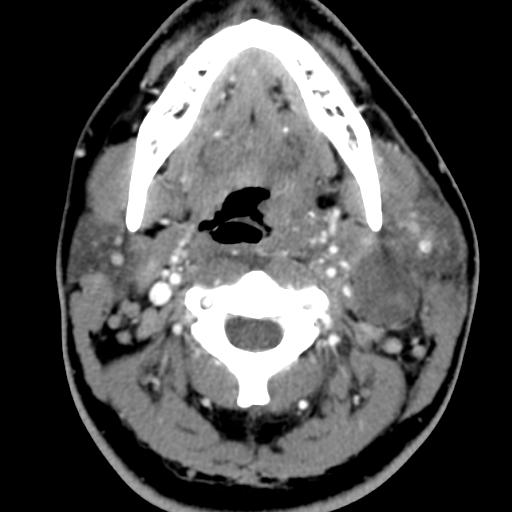
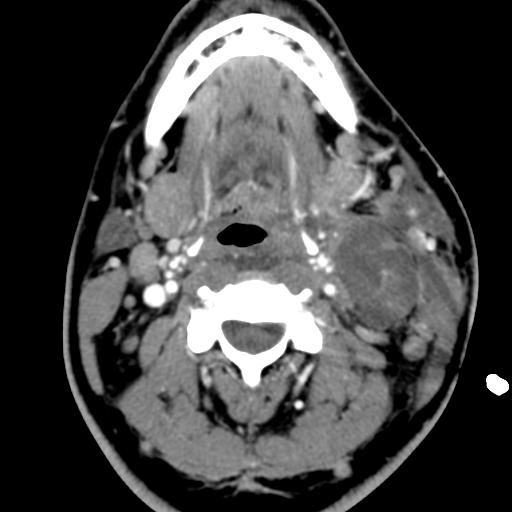
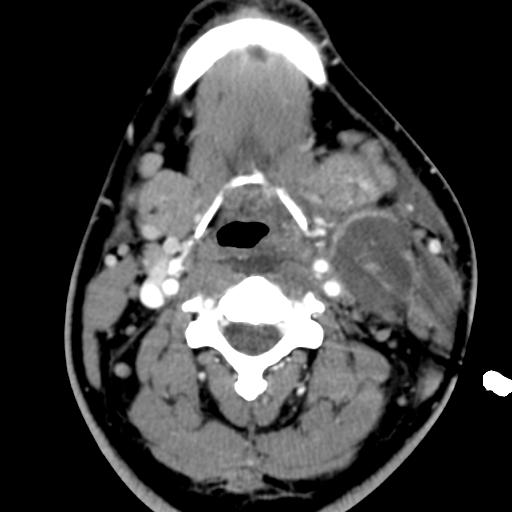
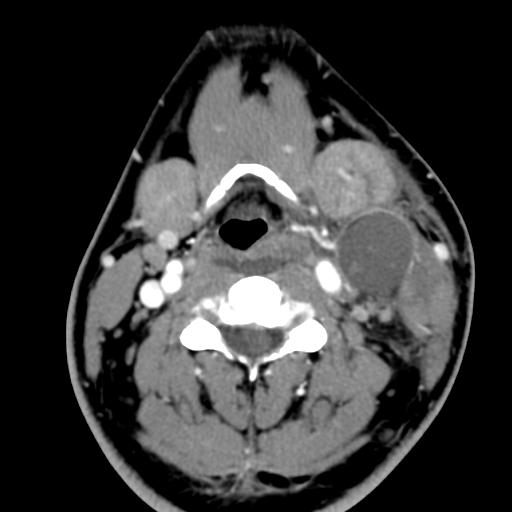
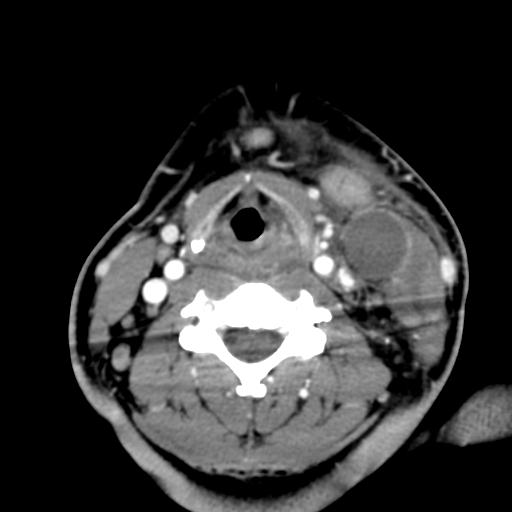
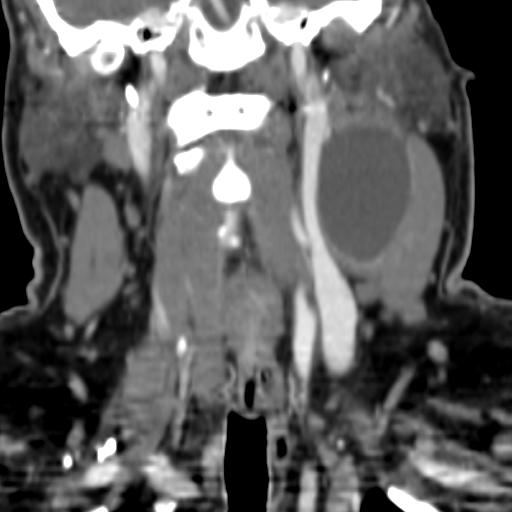
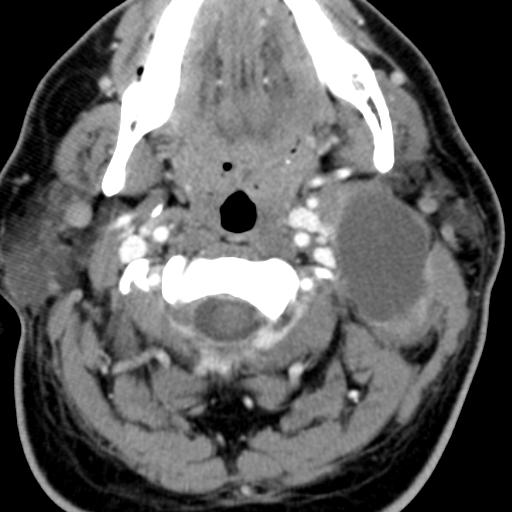
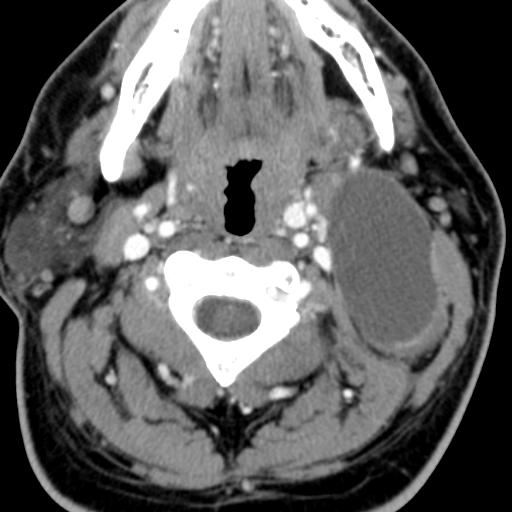
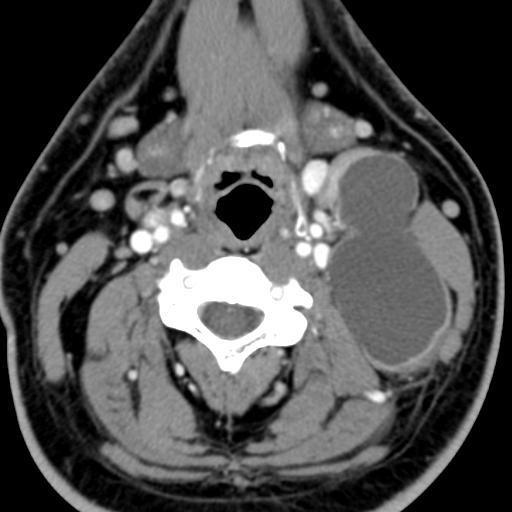
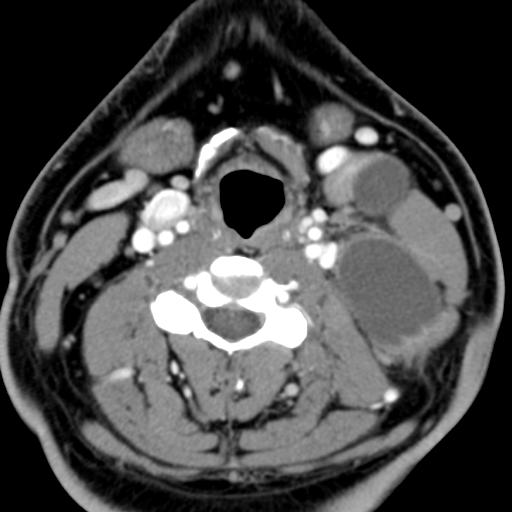
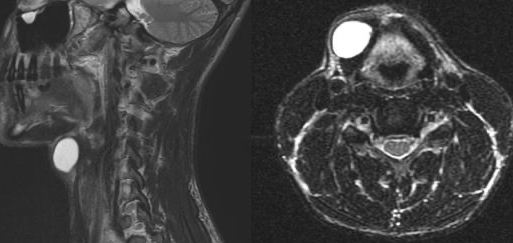
Second branchial cleft anomalies most commonly present as cysts followed by sinuses and fistulae. Most are present within the submandibular space but they can occur anywhere along the course of the second branchial arch tract which extends from the skin overlying the supraclavicular fossa, between the internal and external carotid arteries, to enter the pharynx at the level of the tonsillar fossa. They have previously been classified into four different subtypes by Bailey in 1929:
Type I – Most superficial and lies along the anterior surface of sternocleidomastoid deep to the platysma, but not in contact with the carotid sheath.
Type II – Most common type where the branchial cleft cyst lies anterior to the sternocleidomastoid muscle, posterior to the submandibular gland, adjacent and lateral to the carotid sheath.
Type III – Extends medially between the bifurcation of the internal and external carotid arteries, lateral to the pharyngeal wall.
Type IV- Lies deep to the carotid sheath within the pharyngeal mucosal space and opens into the pharynx.
Case 1-Infected
In adult patients, the main diagnostic consideration is whether the cystic lesion represents a metastatic lymph node and subsequent imaging is directed at identifying a primary neoplastic lesion. This is particularly true if there is no history of chronic neck fullness and no history of a recurrent mass following upper respiratory tract infections. Occult papillary thyroid cancer is also a recognised cause of cystic metastases and may also seen seen in children. Fluid aspiration in association with thyroglobulin levels may aid the distinction.
case 2 infected-treatment.
On ultrasound, second branchial cleft cysts are typically well-circumscribed, thin-walled and anechoic with evidence of compressibility and posterior acoustic enhancement. They may contain internal echoes compatible with internal debris.
Case 3
On CT imaging, they are well-circumscribed, low-density cystic masses with a thin wall. If they become infected, this may become thick-walled with evidence of mural enhancement, localised inflammatory change and perilesional fat stranding. The mural thickening is attributed to the response of lymphoid tissue.
case 4
MRI is better suited in the assessment of deep tissue involvement. On T1-weighted imaging, they may turn from low to high signal depending on the proteinaceous content of the cyst, but are typically hyperintense on T2. As with CT imaging, mural thickening and enhancement varies with inflammatory change and typically occurs in the setting of infection. A tissue ‘beak’ between the internal and external carotid arteries is pathognomonic of Bailey type III cysts. Surgical management involves complete surgical excision encompassing the external sinus opening with dissection of the sinus tract.
case 5
Reference:
Adams, A., Mankad, K., Offiah, C. et al. Branchial cleft anomalies: a pictorial review of embryological development and spectrum of imaging findings. Insights Imaging7, 69–76 (2016). https://doi.org/10.1007/s13244-015-0454-5
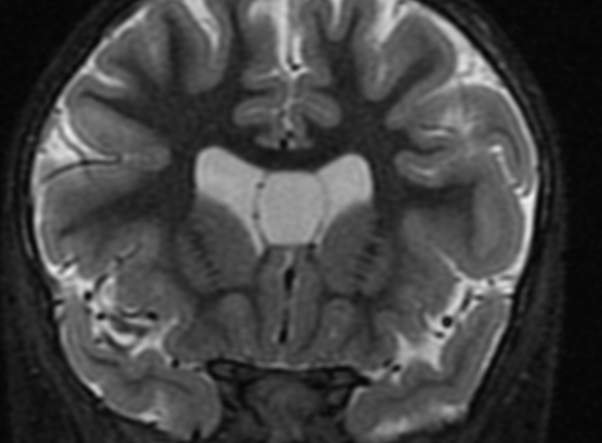
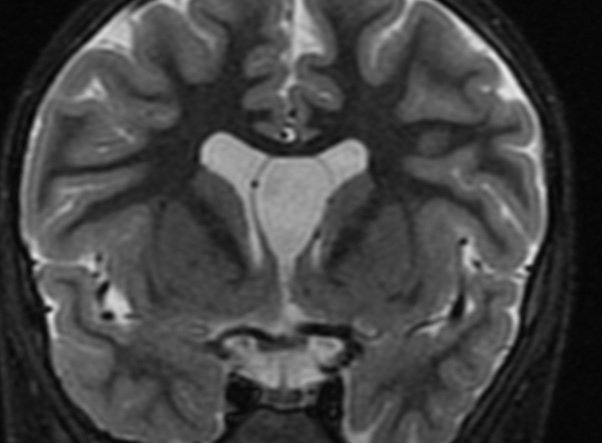
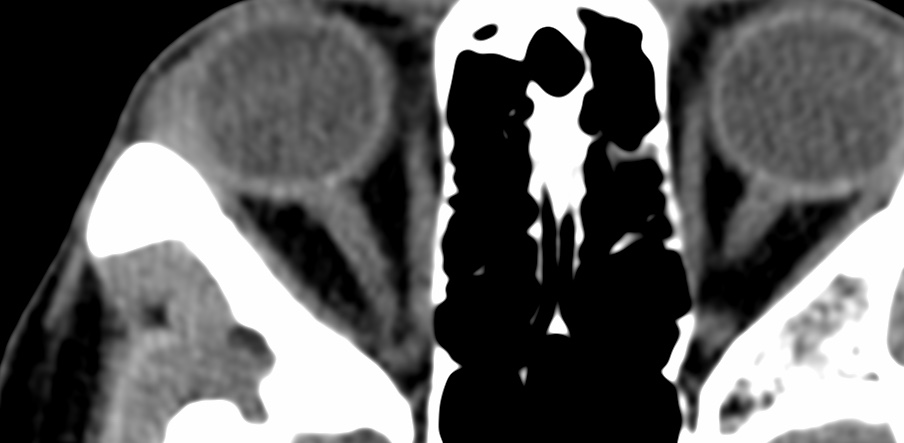
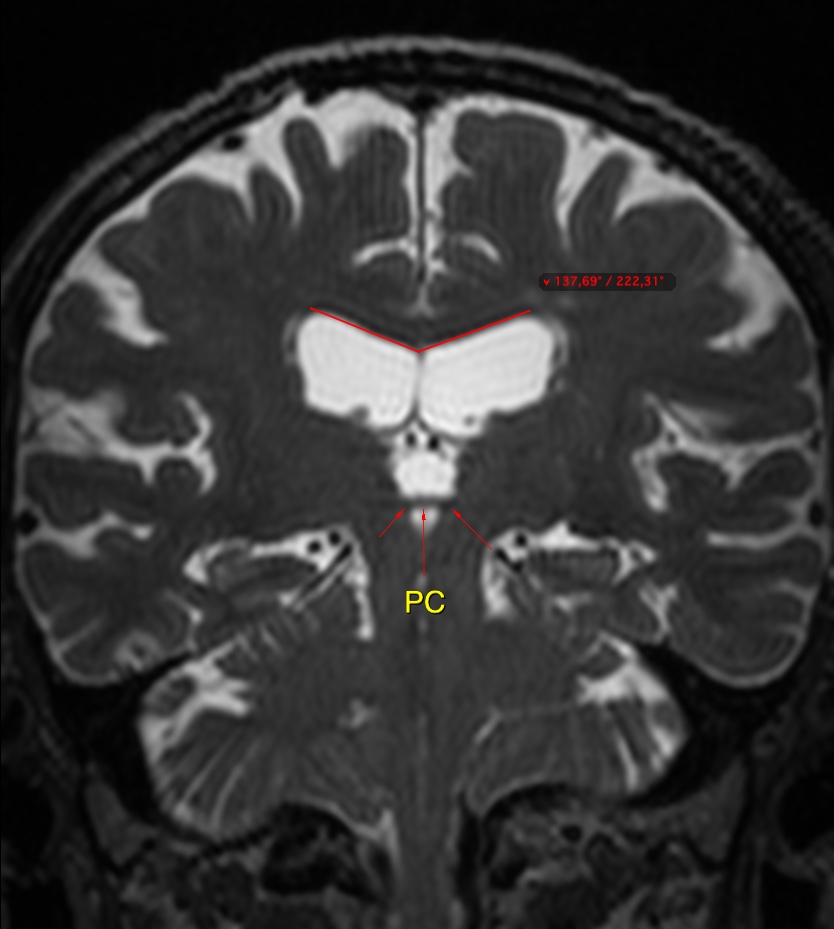
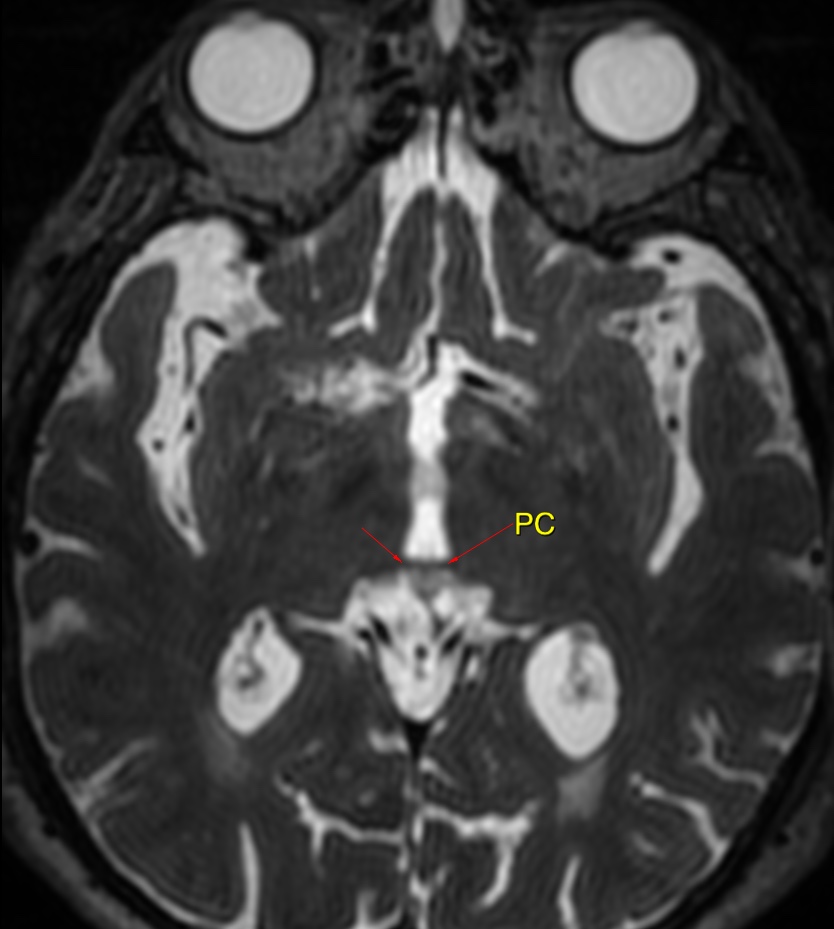
The callosal angle has been described as useful in discriminating iNPH-Hakim–Adams Syndrome from ventricular dilation secondary to atrophy. the angle should be measured on a coronal image perpendicular to the anterior commissure – posterior commissure (AC-PC) plane at the level of the posterior commissure.
A normal value is typically between 100-120°. In patients with iNPH that value is lower, between 50-80°