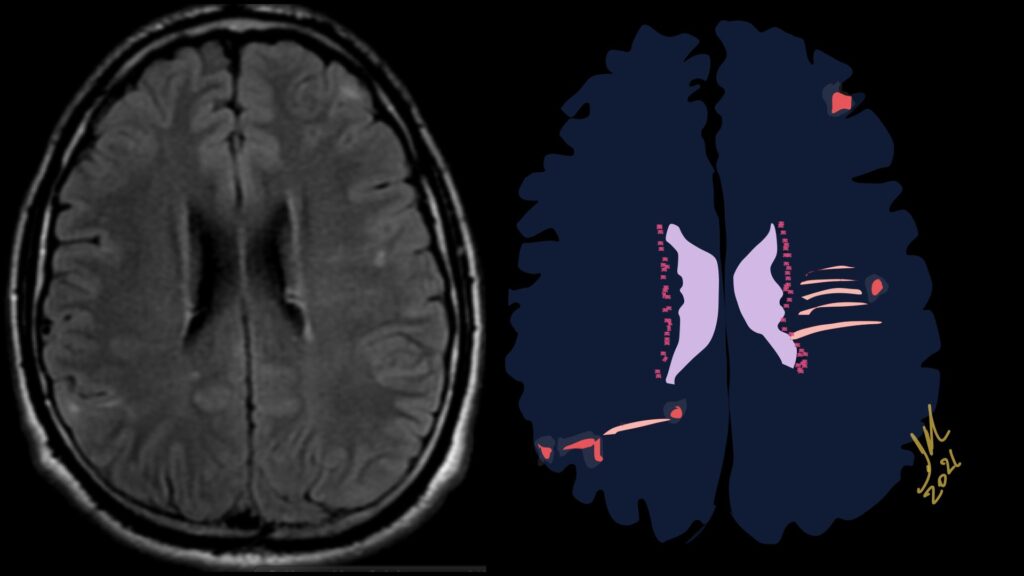
Imaging features, without any physical findings or symptoms, can also be used to make the diagnosis of TS. These unique diagnostic findings include the presence (on CT, MR, US) of multiple subependymal nodules (SENs; especially when calcified) or wedge-shaped cortical calcifications, intraventricular tumor consistent with subependymal giant cell astrocytoma (SGCA), and multiple cortical or subcortical “tubers” (especially if associated with subcortical white matter edema). In addition, typical linear abnormalities in the white matter may be found, Cystic lacune–like lesions have also been described in up to 44% of TS patients.
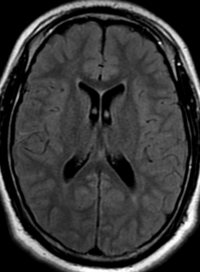
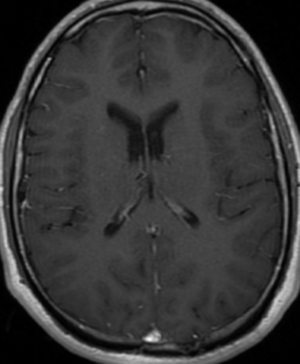
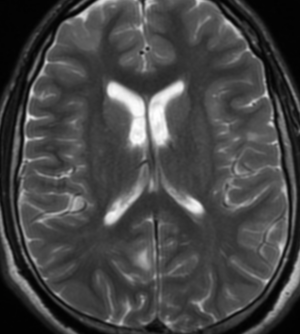
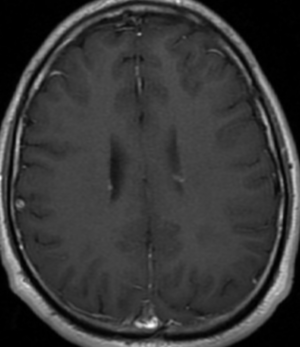
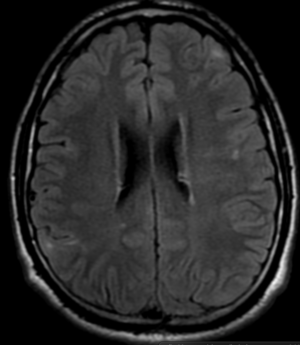
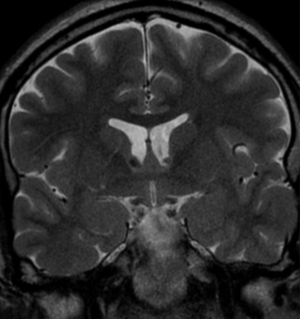
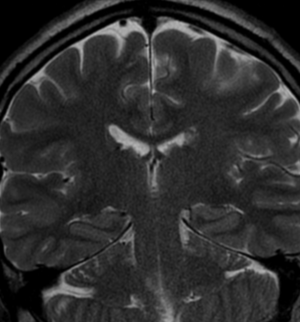
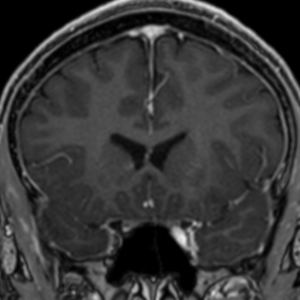
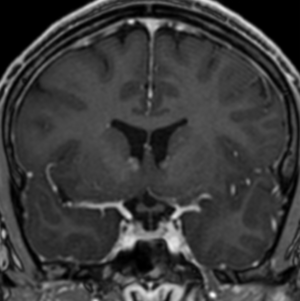
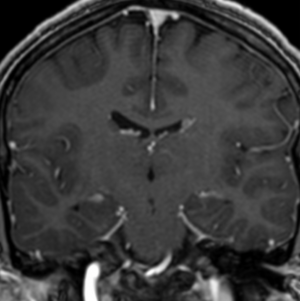
Radial Lines and subcortical tubers and subependymal nodules (SENs).
The SENs, although quite obvious on CT can be subtle on MR. The signal intensity patterns are variable and can change over time. Some SENs may have a “target” appearance on T2-weighted MRI, with a central hyperintensity reminiscent of the gyral core seen in the cortical tubers. the center of a SEN can be bright on the T1-weighted image.This appearance could be related to the paradoxical T1 shortening that occurs with dispersed microscopic calcifications. SENs show gadolinium enhancement, often clustered about the foramen of Monro. The SENs are histologically similar to the cortical hamartomas but are smaller and arise primarily in the striothalamate groove along the lateral margin of the lateral ventricles.
The SENs do not appear to grow; however, they do calcify progressively during the first two decades, so that by age 20 years virtually 100% are hyperdense, This presence of calcification allows SENs to be distinguished from the otherwise similar-appearing subependymal gray matter heterotopias. Heterotopic gray matter is isodense and isointense to normal gray matter, without contrast enhancement
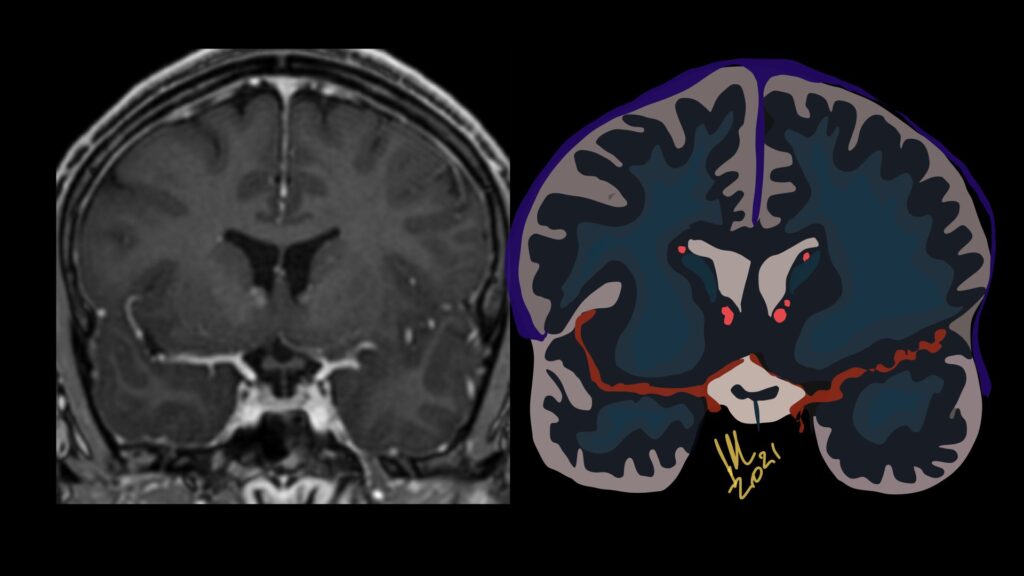
Tuberous sclerosis complex (TSC) or Bourneville-Pringle syndrome is a Multisystem genetic disorder with epilepsy, multiorgan tumors, and hamartomas. Spectrum of central nervous system, hamartomas, all contain dysplastic neurons and giant (balloon) cells. Caused by mutation in TSC1 or TSC2 gene. Now considered infantile (developmental) tauopathy. Tau abnormally expressed in many of dysmorphic neurons and glial cells of TSC.